Clear Sky Science · zh
用于基础机器学习原子间势的图形原子簇展开
教会计算机感知原子
为电池、飞机或聚变反应堆设计新材料,往往归结为一个简单的问题:原子如何相互推拉?精确计算这些力代价极高,对单一材料在超级计算机上可能需要数天时间。本文提出了一类新的机器学习模型,称为 GRACE,它像一个通用“计算器”一样,能够处理元素周期表上大多数元素的原子力。它们的目标是使复杂材料的精确模拟从需要英雄式努力变为日常可行。
一个模型,适用于多种材料
现有的大多数机器学习力场是专用工具:它们在少数元素或化合物上表现非常好,但当加入新元素时必须从头重建。GRACE 采取了不同的路径。它从一开始就被设计为基础模型,能够用一套共享规则处理89种化学元素和极为多样的原子排列。为此,作者以一种称为原子簇展开的数学框架为基础,并将其扩展到图状结构,使模型能够在统一方式下描述局部原子邻域和更广泛的模式。GRACE 不再通过硬编码每一种可能的相互作用,而是学习紧凑的“嵌入”来捕捉元素之间的相似性,从而使对一种材料的知识能够帮助描述另一种材料。
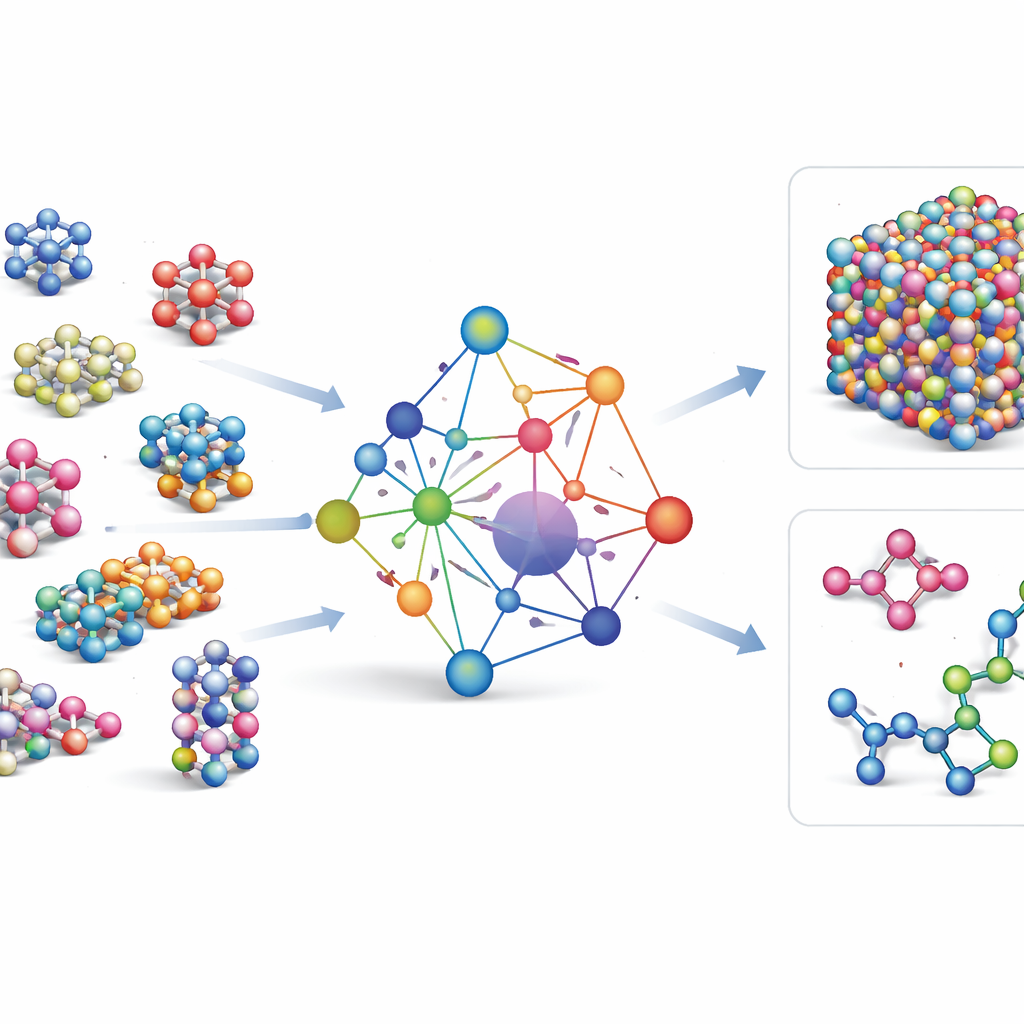
在大量原子数据上训练
为了教会 GRACE 原子的行为,作者汇集了若干最大的公开量子力学计算数据库。核心是 OMat24 集合,包含约1.1亿次无机材料的模拟,辅以另外两个跟踪结构弛豫与演化的数据集。一起,这些数据集覆盖了近平衡晶体、受应变和扭曲的结构、高温液体等多种情况,并且涵盖了相同的广泛元素集合。GRACE 模型有不同规模,从只查看局部原子环境的简化单层版本到通过“消息传递”在相邻区域间有效交流的更深双层版本。初始训练在能量、力和内部应力之间寻求良好平衡,随后通过微调使模型与材料科学中广泛使用的参考数据库兼容。
将模型付诸检验
一个通用模型只有在能可靠地完成多项任务时才有用。因此作者将 GRACE 置于一个严格的测试套件中,模拟科学家实际使用原子尺度模拟的方式。在用于发现稳定晶体结构的社区基准上,GRACE 持续位于“帕累托前沿”:在相同精度下,它比竞争模型更快;在相同速度下,它更精确。预测热导率时也出现类似优势——热导率对原子运动的微小变化极为敏感。GRACE 在弹性性质、表面能、晶界能以及许多纯金属的点缺陷形成能方面也表现良好,这些都检验了材料在拉伸、切割或局部损伤时的响应。对高温熔盐进行的长时间分子动力学运行显示,该模型在数纳秒时间尺度上数值稳定,同时能复现细致的结构模式和原子扩散速率。
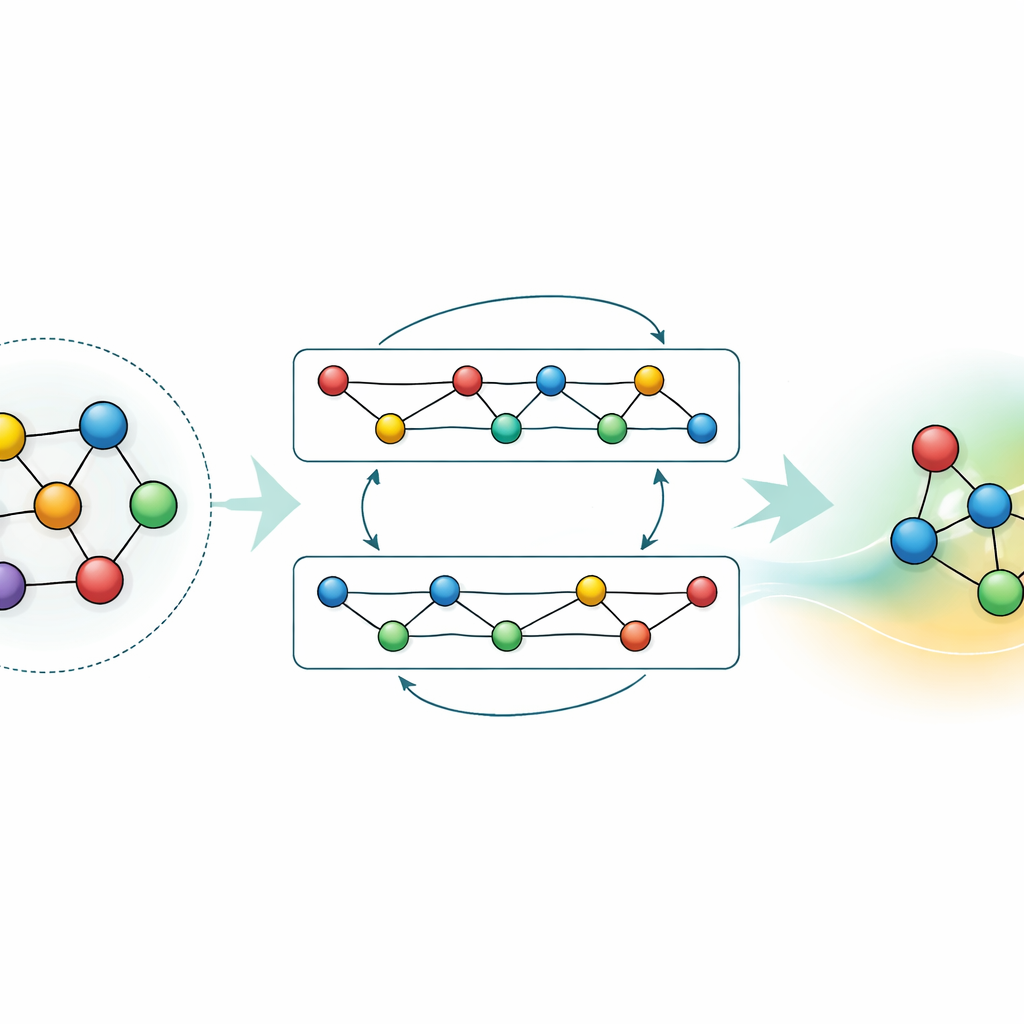
调整与压缩已学到的知识
尽管通用模型很强大,但许多应用要么需要对特定材料更高的精度,要么需要在普通硬件上更快的计算速度。作者展示了两种策略,以在不丢弃 GRACE 已有知识的情况下实现这些目标。首先,他们在聚焦的数据集上对基础模型进行微调,例如铝-锂合金或详细的氢燃烧路径。对于合金,哪怕是适度的额外数据也能显著提高预测精度,优于用相同信息从头训练的模型。对于燃烧问题,直接微调通常会导致模型“遗忘”其他材料的知识;通过谨慎地冻结网络的部分参数并仅更新选定参数,作者在限制灾难性遗忘的同时仍能提升对新化学体系的准确性。其次,他们展示了如何将大型模型蒸馏为更简单的“学生”模型,使其在关键体系上模仿教师模型。该蒸馏版本在 CPU 上运行速度约快70倍,同时保留了大部分精度,尤其在以复杂合金和由原始 GRACE 标注的较简单参考结构混合训练时表现良好。
这对未来材料设计意味着什么
这项工作将 GRACE 定位为下一代原子尺度建模的灵活基础。研究者无需为每种材料或性质从头制作新的势函数,可以从通用的 GRACE 模型出发,然后根据需要进行微调或蒸馏,从而节省大量计算时间和专家工作。基准测试表明,这种方法不仅能匹配现有工具;在速度和可靠性上通常还优于它们,尤其是在像热传输这样苛刻的性质预测上。对于非专业人士,关键信息是:一个精心设计的、通用的机器学习模型现在可以作为覆盖元素周期表大部分区域的可信“引擎”,用于虚拟实验,推进更好电池、催化剂、结构合金和能源材料的发现。
引用: Lysogorskiy, Y., Bochkarev, A. & Drautz, R. Graph atomic cluster expansion for foundational machine learning interatomic potentials. npj Comput Mater 12, 114 (2026). https://doi.org/10.1038/s41524-026-01979-1
关键词: 机器学习原子间势, 材料建模, 原子模拟, 基础模型, 图形原子簇展开