Clear Sky Science · zh
机器学习力场在金属元素间产生误差的起源
为什么某些金属对人工智能更难以理解
机器学习模型正成为模拟原子运动的强大工具,与传统的量子计算相比可为科学家节省大量计算时间。你可能会认为最简单的材料——由单一元素构成的纯金属——应当是这些模型最容易学习的对象。但这项研究显示事实并非如此:一些金属仍然很难被准确描述,作者们找到了一个物理学上的原因。
构建一个大而干净的金属行为图谱
为系统地研究这个问题,研究人员创建了一个名为 Metal-43 的新数据集,基于严格的量子力学计算。它涵盖了 43 种不同的金属元素,从轻质的锂到重金属钨,全部在一致的计算设置下处理。对于每种金属,他们在若干温度下模拟了成千上万种原子结构,记录每个原子的能量和力。这个经过精心控制的“游乐场”让他们可以在公平、可比的条件下,在许多金属上测试机器学习力场——即预测原子力的 AI 模型。 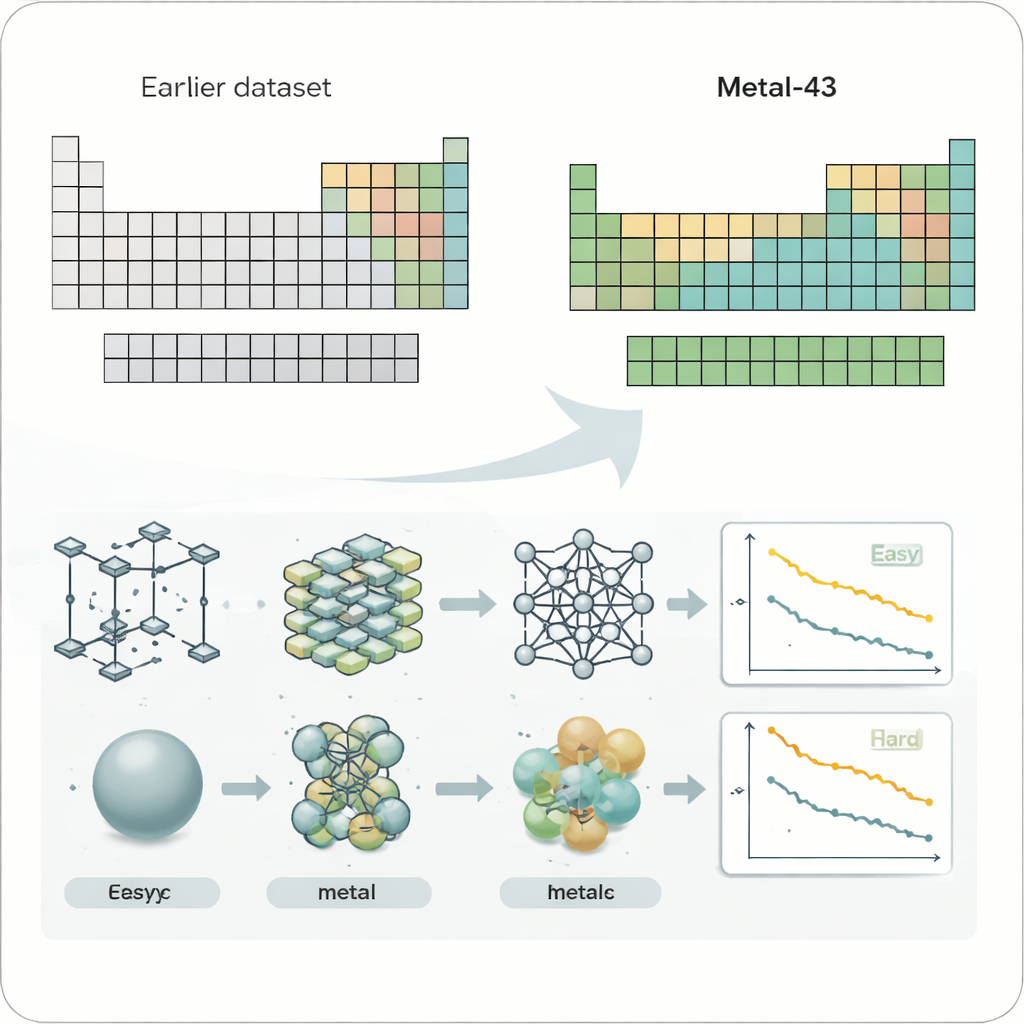
模型误差如何在元素周期表上排列
研究考察了四种广泛使用的机器学习力场模型,包括为每种元素分别训练的紧凑模型以及在许多体系上同时训练的大型通用模型。当作者把预测误差在元素周期表上绘制出来时,出现了一个显著的模式。诸如碱金属和碱土金属等结合较弱的软性金属对所有模型而言通常较容易,而周期表中部的早期过渡金属——那些常用于高性能合金和催化剂的元素——则持续表现出更大的误差。即便在将原始误差按原子力的总体强度重新缩放后,这一趋势仍然存在,表明困难不仅仅是键更强的问题,而是更根本的原因。
金属“电子交通图”中隐藏的复杂性
这项工作的关键见解是将这些模型误差与每种金属的费米面形状联系起来,费米面是一种三维的“交通图”,表示电子在最关键能量处可以如何移动。在易于拟合的金属中,这个面光滑且接近球形。而在难以拟合的早期过渡金属中,它变得参差不齐、出现口袋状结构,反映出与部分填充的 d 轨道相关的复杂电子行为。当原子被扰动或轻微位移时,这些复杂的费米面会以不均匀、时而突变的方式改变,进而使整体能量景观变得粗糙且复杂。作者展示了某些电子能量总和在小扰动下波动速度的简单数值度量与机器学习误差大小之间存在强相关性,尤其是对于那些有问题的过渡金属。
即使在理想化数据上,当前 AI 模型的局限
为了把金属自身的困难与当前 AI 方法的局限区分开,团队还使用传统的人工构造原子力模型生成了人工数据集。其中一些旧模型主要依赖原子间距离,而另一些则包含强烈的角度依赖以模拟更具方向性的键合。机器学习力场几乎可以完美地再现基于距离的模型,但在角度效应重要时其误差急剧增加——特别是对于那些已知难处理的金属。该比较表明,挑战不仅在于金属的底层物理,还在于当今机器学习架构的表现能力,它们在处理强角度依赖的多体相互作用时仍然存在困难。
这对未来模拟的意义
对非专业读者而言,主要结论是有一个明确且有物理依据的原因解释为何某些金属比其他金属更难被 AI 模拟:它们在费米能级处的电子运动复杂性使能量景观变得粗糙而精细。Metal-43 数据集和本文提出的简单电子结构指标为研究者提供了一种预测哪些材料将成为棘手对象、对新模型进行公平基准测试以及设计能更好捕捉方向性键合的改进型力场的方法。从长期看,这些见解有助于使基于 AI 的模拟在设计先进合金、催化剂和其他金属相关技术时更可靠。
引用: Geng, X., Zhang, W., Wang, LW. et al. Origin of the machine learning forces field errors across metal elements. npj Comput Mater 12, 102 (2026). https://doi.org/10.1038/s41524-026-01977-3
关键词: 机器学习力场, 金属材料, 费米面, 原子间势, 密度泛函理论