Clear Sky Science · zh
JAK/STAT1-干扰素-ISGylation 网络在乳腺癌对 FOXM1 和 CDK4/6 抑制剂耐药中的作用
这对乳腺癌治疗为何重要
许多雌激素受体阳性(ER+)乳腺癌患者现在接受能够通过阻断细胞周期来减缓肿瘤生长的现代靶向药物。然而,肿瘤几乎不可避免地会学会绕过这些治疗并再次生长。本研究提出了一个紧迫的问题:当 ER+ 乳腺癌对两类主要药物——FOXM1 抑制剂和 CDK4/6 抑制剂——产生耐药时,癌细胞内部发生了哪些改变使它们能够逃逸,这些改变是否也能揭示阻止它们的新方法?
耐药肿瘤内的共同生存线路
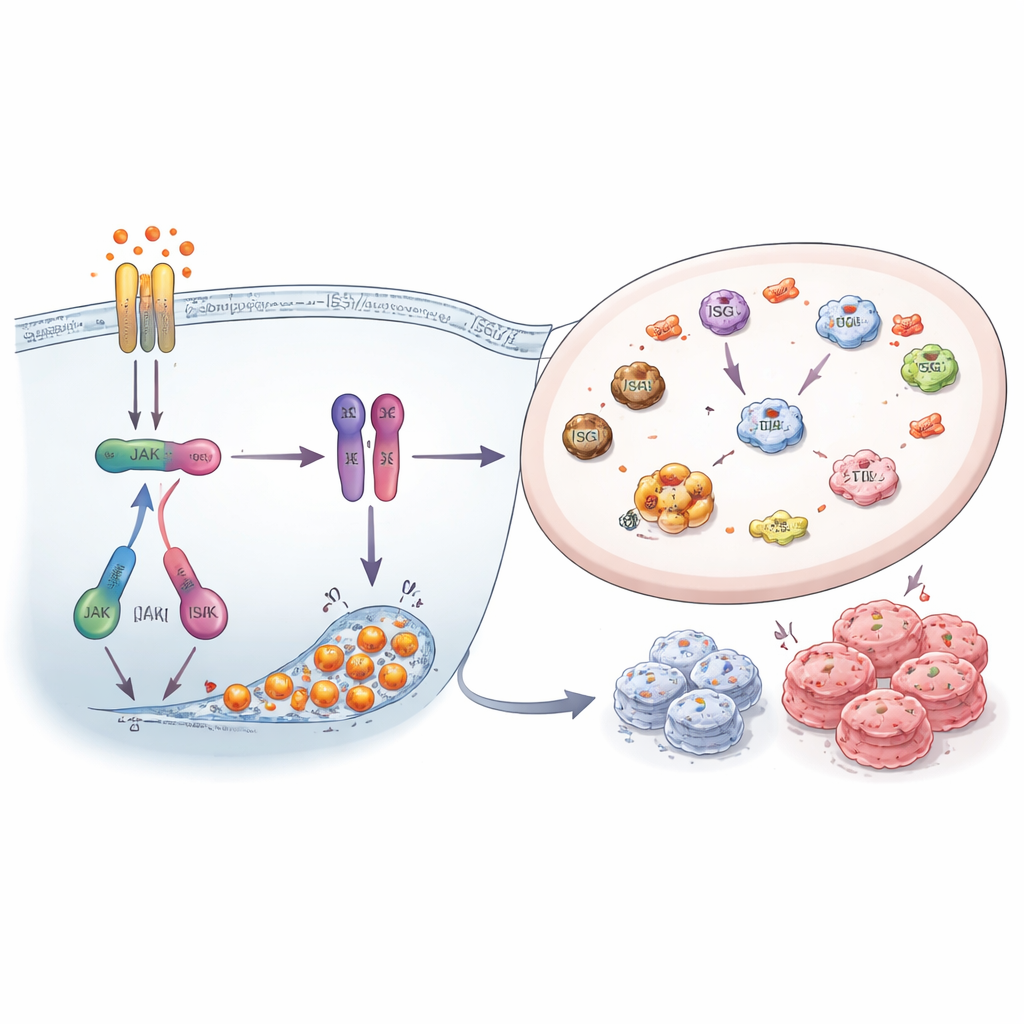
研究人员聚焦于在体外培养直到对 FOXM1 抑制剂或对如帕博西尼(palbociclib)和阿贝昔替尼(abemaciclib)等 CDK4/6 抑制剂不再响应的 ER+ 乳腺癌细胞。他们发现,尽管这些细胞对不同药物产生了耐药性,但它们启动了类似的内部警报系统,该系统以干扰素信号和名为 STAT1 的蛋白为核心。该警报导致大量“干扰素刺激基因”(ISGs)和一种称为 ISG15 的小蛋白的产生,ISG15 可以像分子标签一样连接到其他蛋白上。与非耐药细胞相比,耐药细胞中 STAT1、激活的 STAT1、游离 ISG15 及 ISG15 标记的蛋白水平明显升高,表明该网络构成了药物耐受性的共同骨架。
一种保护性的分子标签外衣
更细致地观察表明,耐药细胞不仅产生更多 ISG15,还上调了将 ISG15 连接到其他蛋白上的酶,即 ISGylation 过程所依赖的酶。这些酶——HERC5、HERC6 和 UBE2L6——在耐药细胞中显著升高,尤其是在对 FOXM1 抑制剂耐药的细胞中。包括 STAT1 本身在内的许多细胞蛋白在耐药细胞中带有 ISG15 标签,且往往高于原始的药物敏感细胞。由于添加这些标签可以改变蛋白质的稳定性和功能,ISGylated 蛋白的积累似乎是癌细胞增强治疗耐受性的一个组成部分。
关闭警报使耐药细胞变脆弱
研究者接着探究是否抑制该警报网络会使耐药细胞更易受攻击。他们使用阻断 JAK 激酶的药物——JAK 是 STAT1 上游的关键开关——以及小干扰 RNA 来降低 ISG15、HERC5 和 HERC6 的表达。阻断 JAK1/2 信号显著降低了 STAT1 活性,减少了 ISG15 水平和 ISGylation,并缩小了耐药细胞形成的克隆,尤其是对 FOXM1 抑制剂耐药的细胞。同样,直接沉默 ISG15 及其标记酶减少了总体的 ISGylation 模式并削弱了细胞存活。这些实验表明,干扰素–STAT1–ISG15 系统不仅是旁观者,而是积极支持耐药癌细胞生长与持久存在的机制。
序贯治疗策略带来新希望
最令人鼓舞的发现之一是,耐药细胞并非陷入单一不可战胜的状态。对 CDK4/6 抑制剂产生耐药性的乳腺癌细胞仍对 FOXM1 抑制剂敏感,而对 FOXM1 抑制剂耐药的细胞仍可被帕博西尼或阿贝昔替尼减缓。在二维细胞层和更能模拟肿瘤的三维基质胶(Matrigel)培养中,改变药物类别能显著降低细胞生长并下调驱动 DNA 复制和细胞分裂的基因表达。同时,患者数据表明,ER+/HER2− 肿瘤中较高的 ISG15 及其相关酶的表达与更差的生存期相关,强调了该耐药线路的临床相关性。
这对患者与未来疗法的意义
对外行读者而言,研究描绘的图景是:耐药的 ER+ 乳腺癌通过围绕一个共同的应激反应回路重塑自身,利用干扰素信号和 ISG15 标签作为某种保护性装甲。好消息是,这层装甲暴露了新的薄弱环节。阻断 JAK–STAT1–ISG15 通路的药物,以及在 CDK4/6 药物之后(或反之)序贯使用 FOXM1 抑制剂的治疗方案,可能有助于超越耐药而不是被其击败。尽管这些发现仍需在临床试验中验证,但它们为将一种常见的治疗死胡同转化为新的控制机会提供了更清晰的路线图。
引用: Ziegler, Y., Kumar, S., Saeh, C.M. et al. JAK/STAT1-interferon-ISGylation networks in breast cancer resistance to inhibitors of FOXM1 and CDK4/6. npj Breast Cancer 12, 44 (2026). https://doi.org/10.1038/s41523-026-00911-6
关键词: 雌激素受体阳性乳腺癌, 药物耐药, CDK4/6 抑制剂, FOXM1 抑制剂, 干扰素信号传导