Clear Sky Science · zh
用于大规模光电性质预测的物理启发哈密顿学习
这对更高效的太阳能电池和发光二极管为何重要
设计下一代太阳能电池、LED 及其他基于光的技术越来越依赖于模拟电子在复杂材料中的运动。但最精确的量子力学计算在计算量上非常沉重,面对含有数万原子的真实、有序或无序晶体时往往难以胜任。本文提出了一种名为 HAMSTER 的新方法,将成熟的物理模型与机器学习相结合,使这些大尺度、现实的模拟既可行又可靠。

一种仍尊重物理的捷径
工作的核心是如何预测哈密顿量——这一描述材料中电子行为的关键数学对象。如果知道哈密顿量,就能计算带隙等决定材料如何吸收和发射光的重要量。纯数据驱动的神经网络可以从原子位置映射到哈密顿量,但通常需要庞大的训练集且难以解释模型内部机理。作者改为从一个公认的近似物理模型——紧束缚模型出发,该模型已能捕捉原子间的主要相互作用。机器学习部分仅需学习该近似与高精度量子计算之间的残差,大幅降低了学习负担。
教模型“感受”周围环境
一个关键创新是 HAMSTER 如何对每对原子的“环境”进行编码。在真实材料中,随着温度升高原子会振动和移动,邻近原子会微妙地改变一对给定位点之间电子的跃迁。传统紧束缚模型在很大程度上忽略了这些多原子影响。HAMSTER 用一个紧凑的描述符表示两相互作用原子的局部环境,该描述符反映哪些邻居在选定距离内、它们与目标原子的距离以及轨道的取向。平滑的截断函数确保远处原子贡献较小。随后,一个简单的径向基机器学习模型使用这些描述符对紧束缚哈密顿量元添加小的修正,集中处理缺失的环境效应,而不是从头重学基本物理。
从简单半导体到复杂钙钛矿
为验证这一思想,研究团队首先将 HAMSTER 应用于砷化镓——一种研究充分的半导体,结果显示仅用少量训练结构就能在预测能级方面达到接近第一性原理的精度。随后他们挑战更困难的目标:卤化物钙钛矿(如 CsPbBr3 和 MAPbBr3),这些材料在太阳能和发光器件中很有前景,但由于晶格软且热涨落强烈而难以建模。对于 CsPbBr3,使用单一温度下的分子动力学快照训练的 HAMSTER 能在宽广温度范围内重现精细的量子计算结果,使带隙和能级误差保持在几十毫电子伏以内。它还捕捉到随着原子运动带隙随时间波动的特性,这是进行现实器件预测的关键因素。
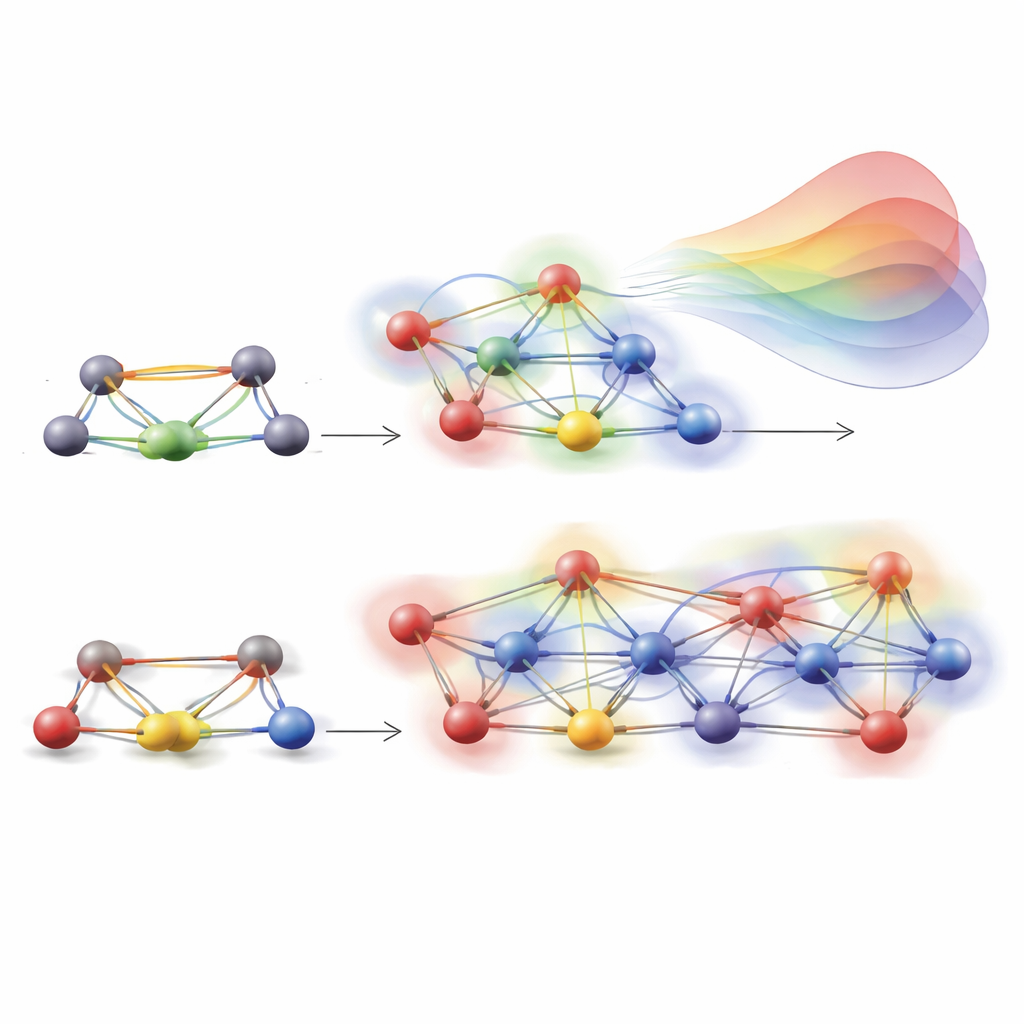
达到真正的大尺度系统
由于 HAMSTER 相对于完整量子计算成本低得多,作者能够扩大到包含数万原子的模拟盒——这是常规模拟所无法企及的尺度。对于 CsPbBr3,他们将用于原子运动的机器学习力场与用于电子结构的 HAMSTER 结合,分析了一个 16 × 16 × 16 超胞,包含超过 20,000 个原子。在这些巨大的系统中,短期的带隙波动相互抵消,显现出与实验测量良好一致的清晰温度趋势。对 MAPbBr3 采取类似策略则使他们能研究接近 50,000 个原子的晶胞,并绘制出体系尺寸与温度如何共同影响带隙的图景,定性上同样与实验相符。
这对未来材料设计意味着什么
总体而言,这项研究表明将物理知识融入机器学习是一条有力路径,可弥合简化模型与完全第一性原理模拟之间的鸿沟。HAMSTER 保留了基于哈密顿量描述的可解释性,同时实现了处理热效应、化学替代和现实尺度所需的精度与通用性。对于非专业读者来说,结论是:这种物理引导的学习方法有望成为在计算机上探索新型光伏与发光材料的实用工具,在不承担传统量子计算高昂代价的前提下,引导实验朝最有希望的候选材料迈进。
引用: Schwade, M., Zhang, S., Vonhoff, F. et al. Physics-informed Hamiltonian learning for large-scale optoelectronic property prediction. Nat Commun 17, 2652 (2026). https://doi.org/10.1038/s41467-026-70865-7
关键词: 卤化物钙钛矿, 材料科学中的机器学习, 电子结构, 光电性质, 紧束缚哈密顿量