Clear Sky Science · zh
从力与噪声中学习数据高效的粗粒化分子动力学
为什么缩小分子重要
模拟蛋白质及其周围水分子中每个原子的不断运动,是我们理解生命在分子尺度上如何运作的最佳工具之一。但这些全原子模拟计算量极大,要跟踪蛋白质在生物学相关时间尺度内的折叠、解折或与伴侣相互作用,可能需要在超级计算机上耗费数月时间。本文介绍了一种新的方法,用于构建快速的简化蛋白质模型,这些模型在行为上仍然类似于其全原子对应体,同时所需的训练数据和计算资源远少于以往。
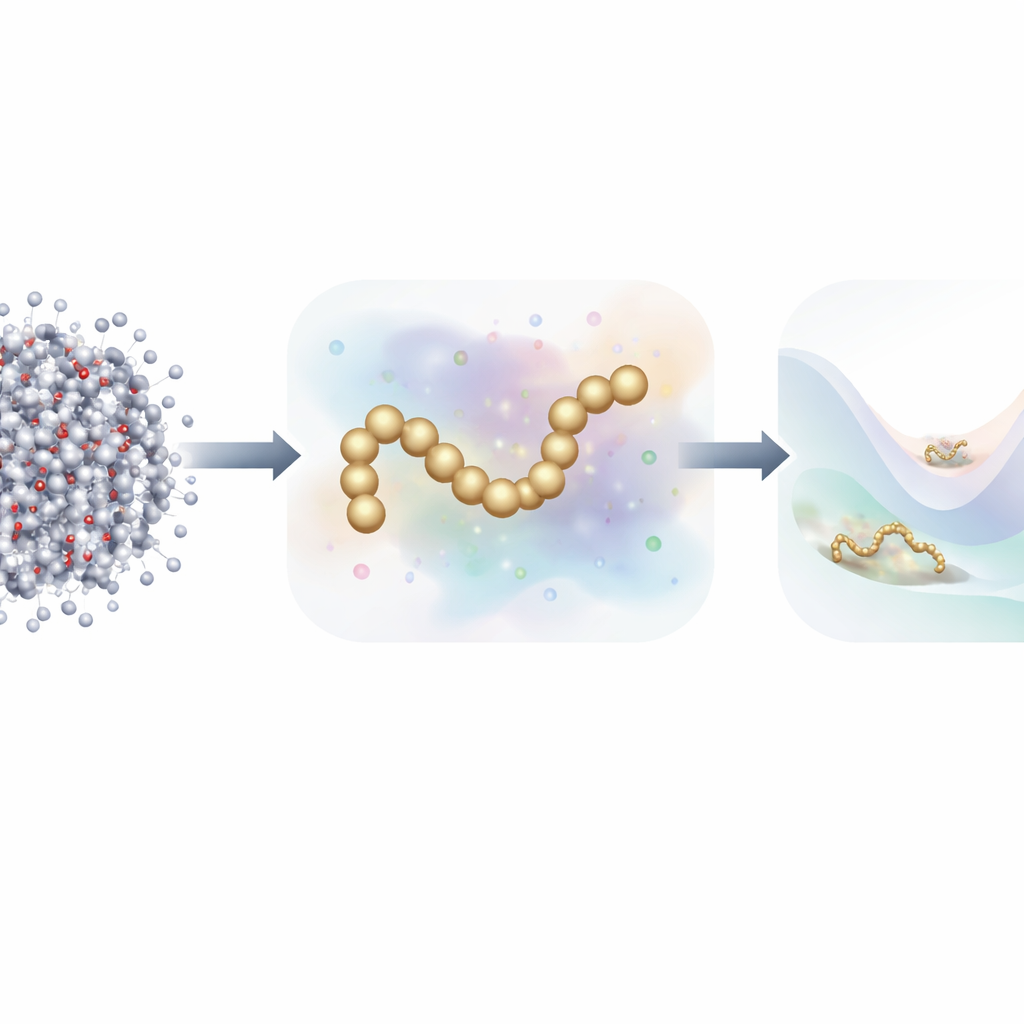
从每个原子到更简洁的图景
传统的分子动力学跟踪每个原子,并在每个极短时间步长上计算它们之间的力。为了加速,科学家常使用粗粒化模型,将许多原子分组为较少数量的“粒子”。这些简化模型运行快得多,但在历史上难以匹配全原子模拟的精度,尤其是对于具有复杂折叠行为的蛋白质。近期工作转向机器学习,以自动发现更好的粗粒化力场,但训练这些模型通常需要数百万个带有每个原子受力标签的详细快照——这是巨大的数据和计算负担。
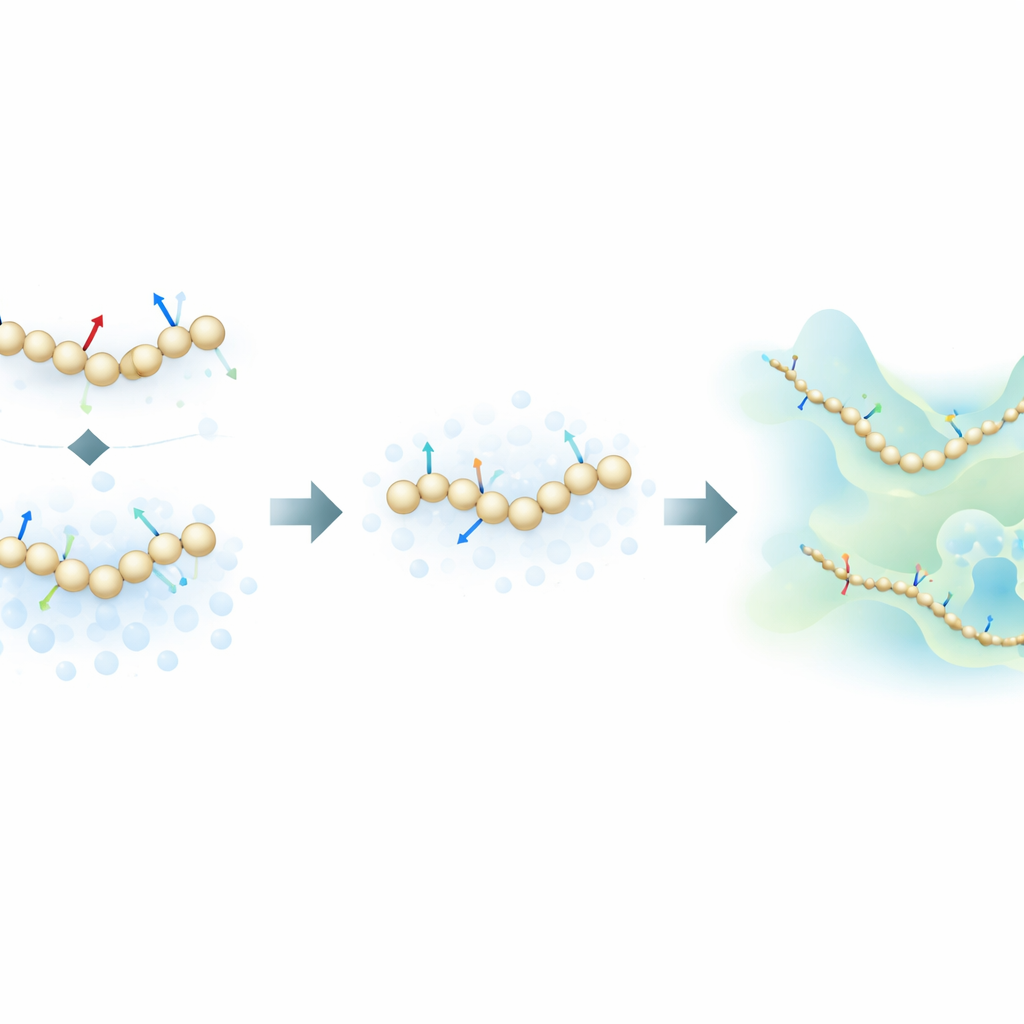
将物理力与有信息的噪声结合
作者提出了一种新的训练策略,借鉴生成式扩散模型的思想——同一类算法也被用于许多现代 AI 图像生成器。与其只从全原子模拟中计算得到的物理力中学习,他们的方法还通过向粗粒化构型故意添加受控噪声来学习分子结构在空间中的分布。在该框架中,噪声不再只是需要被去除的干扰;它成为额外的信息来源。通过在数学上将传统的“力匹配”方法与扩散模型中的去噪技术统一起来,该方法能够用更少的带标签样本推断出蛋白质的底层能量景观。
教会简单模型模仿复杂蛋白质
为检验他们的想法,研究者为几种复杂度逐渐增加的蛋白质训练了神经网络粗粒化模型:小型微蛋白 Chignolin 和 Trp-Cage、稍大一些的 NTL9,以及含 76 个残基的泛素(Ubiquitin)。他们比较了三种训练模式:仅使用原子力、仅使用基于噪声的信息,以及两者结合。对于较小的蛋白质,结果显示新的组合方法能够再现折叠景观的关键特征——例如折叠态与未折叠态的相对稳定性以及中间态的存在——所需的训练数据比标准力匹配方法少多达一百倍。令人惊讶的是,在数据稀缺的情形下,仅用噪声信息训练的模型往往也能匹配或超过仅用力训练的准确性。
扩展到更大、更困难的蛋白体系
泛素是更具挑战性的测试:在真实温度下捕捉其折叠与解折过程历来需要专用硬件和极长的全原子模拟。在此,作者使用一份适中数据集来训练粗粒化模型,数据包括围绕折叠态的短平衡模拟以及强制拉伸蛋白的非平衡“拉伸”模拟。尽管训练集有偏并且缺少在相同条件下的完美全原子参考,结合力与噪声训练的模型仍然恢复了折叠与未折叠态并存的现实图景——且折叠态在稳定性上占优。相比之下,仅用力训练的模型根本无法稳定折叠态,而仅用噪声训练的模型偏好未折叠结构。值得注意的是,任何粗粒化模型都没有简单地记忆训练数据中的极端拉伸构象,这表明学得的能量景观在物理上有意义,而不仅仅是输入轨迹的印记。
这对未来模拟意味着什么
通过将噪声变为训练信号并将其与物理力合并,这项工作表明可以用远比以往想象的更小、更不理想的数据集构建准确的蛋白质粗粒化模型。实际意义是,研究者可能不再需要在专用超级计算机上进行毫秒级的全原子模拟,才能用机器学习的粗粒化动力学探索生物分子的行为。相反,在广泛可用的硬件上进行更温和的模拟就可能足够训练出能够捕捉关键折叠途径和热力学平衡的强大简化模型。尽管关于如何最佳选择与解释所加噪声以及该方法在更大、更复杂生物分子组装上的表现仍有待探讨,但这一方法大幅降低了将数据驱动的粗粒化模拟作为分子科学常规工具的门槛。
引用: Durumeric, A.E.P., Chen, Y., Pasos-Trejo, A.S. et al. Learning data-efficient coarse-grained molecular dynamics from forces and noise. Nat Commun 17, 2493 (2026). https://doi.org/10.1038/s41467-026-70818-0
关键词: 粗粒化分子动力学, 机器学习力场, 蛋白质折叠模拟, 化学中的扩散模型, 数据高效模拟