Clear Sky Science · zh
DiNovo 通过镜像蛋白酶与深度学习实现高覆盖率与高置信度的 de novo 肽测序
以新的细节观察蛋白质
蛋白质是维持细胞活力的微小机器,但要完整读取它们的构件仍然出乎意料地困难。本文介绍了 DiNovo,一款新的软件系统,能比以往更全面、更可靠地“读取”蛋白片段。通过将巧妙的生化策略与现代人工智能结合,它有望发现传统方法常常遗漏的隐匿蛋白、疾病标志物乃至免疫靶点。
为什么读取蛋白片段如此困难
当今大多数蛋白质分析依赖将蛋白切成更小的片段,称为肽段,然后在质谱仪中称量它们的碎片。根据这些质量,计算机试图重建原始肽序列,就像从零碎线索中解填字游戏。现有方法通常假定肽段来自已知的蛋白数据库,这对熟悉的蛋白有效,但在面对新的或意外的蛋白时却力不从心。所谓的 de novo 测序通过直接从数据中读取肽段来避免这个限制,但常常因为某些碎片缺失或肽段并未被干净切割而表现欠佳。
用镜像酶填补空白
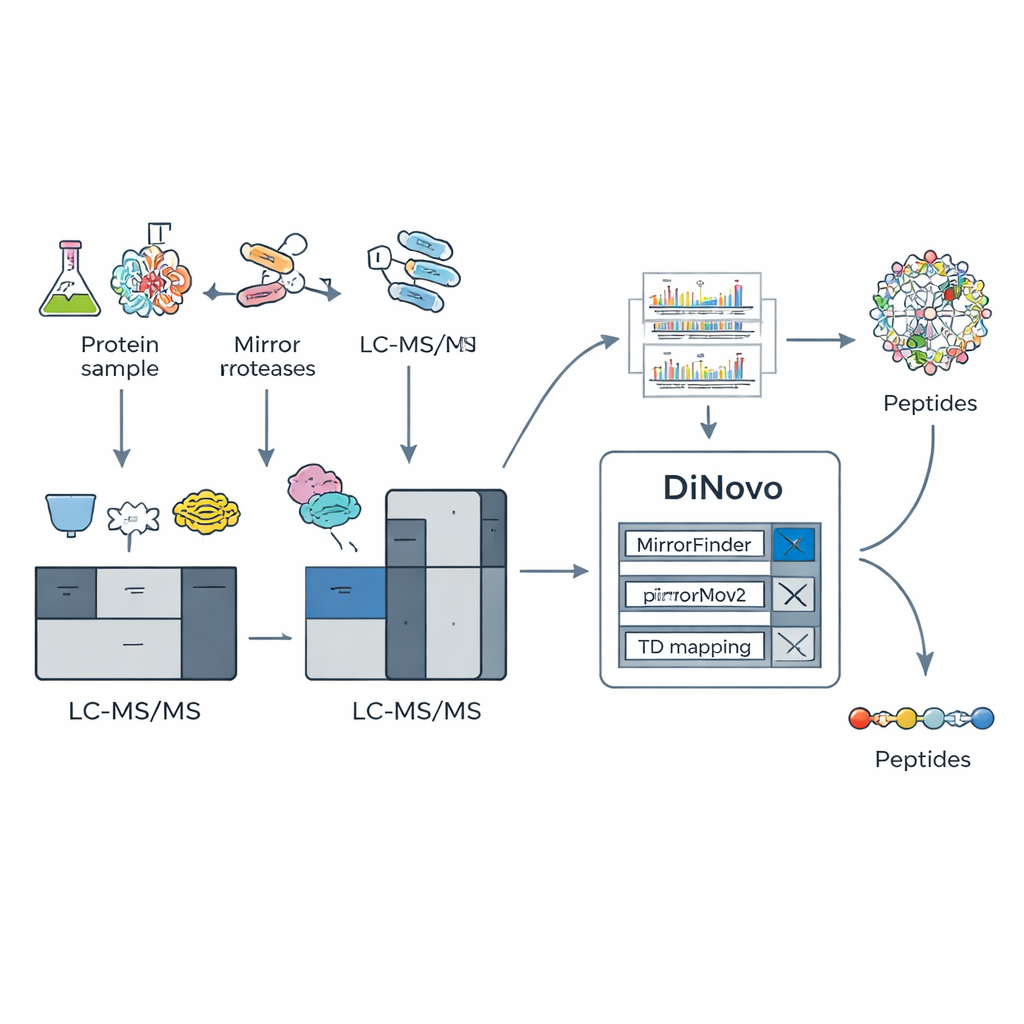
DiNovo 的关键思路是使用成对的“镜像蛋白酶”——成对的切割酶分别在同一类氨基酸的相反两侧切割。例如,一个酶在赖氨酸之前切割,而它的配对酶在该赖氨酸之后切割。这会产生两个相关的肽段,它们共享相同的内部片段但末端不同。当分析这些“镜像”肽段时,它们的质谱包含互补的碎片模式:在一个谱图中缺失的信号常在另一个谱图中出现。作者展示了,结合此类镜像对可以将碎片覆盖率推高到接近完整,约 98% 的可能切点由真实实验信号支持,远高于单一酶切的情况。
为镜像数据量身打造的智能软件流程
为了利用这一生化技巧,团队将 DiNovo 构建为端到端的软件工作流。首先,来自细菌和酵母的蛋白使用两对镜像酶消化,得到的肽段通过高分辨率质谱分析。DiNovo 然后使用一个名为 MirrorFinder 的模块自动识别哪些谱对来自镜像肽段,这一识别直接基于信号模式而非任何预先的序列猜测。接着,其主要的 de novo 引擎 MirrorNovo 使用深度学习来解释这些成对谱图,而备用的基于图的引擎 pNovoM2 则提供了更快的纯 CPU 选项。组合使用这些工具可将谱峰翻译为氨基酸序列,并对那些未形成明显配对的单谱图进行逐一分析,尽可能挖掘信息。
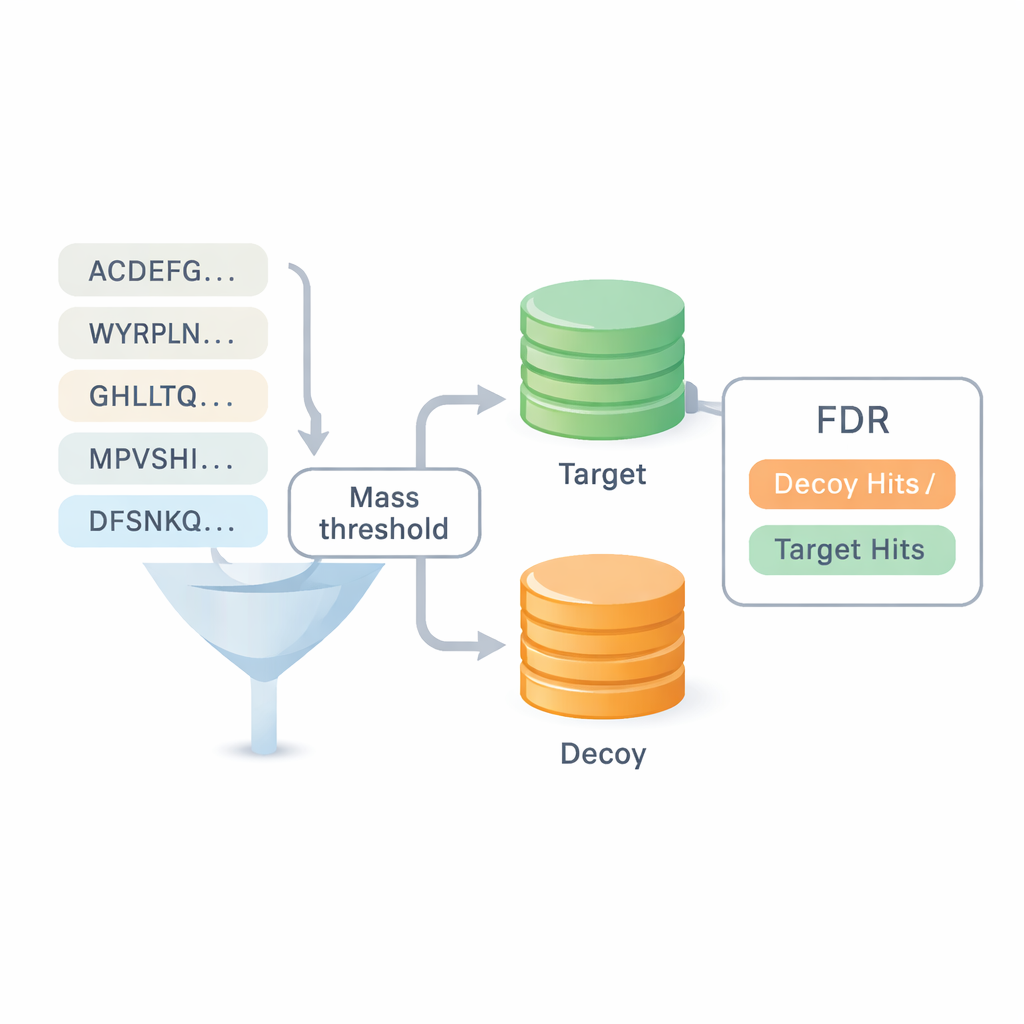
在不依赖旧数据库的情况下衡量可信度
de novo 测序面临的最大问题之一是结果应当被信任到何种程度。大多数现有基准重复使用数据库搜索的答案,这会模糊两种方法之间的界限并掩盖错误。DiNovo 引入了一种不同的质量检验方法,称为目标-诱饵映射。在这里,新读取的肽段被映射到一个由真实(目标)和人工扰乱(诱饵)蛋白序列组成的合并集合。通过比较肽段落入真实集合与扰乱集合的频率,软件可以在不依赖先前鉴定的情况下估计错误率或假阳性率。这使得在相同错误控制下能够将 DiNovo 与标准的数据库搜索程序直接比较。
DiNovo 在实践中的表现
在细菌、酵母和抗体样本的测试中,DiNovo 一贯读取到比仅使用单一酶的著名 de novo 工具更多的肽段和氨基酸。使用两对镜像酶,它产生的高置信度氨基酸数量是经典仅用胰蛋白酶设置的 2–3 倍,并在相似的错误水平下鉴定出更多蛋白。当与三种领先的数据库搜索引擎直接比较时,DiNovo 找到的氨基酸和蛋白数量相近,且其大多数序列在相同谱图上的结果与搜索引擎一致。作者认为,这样的覆盖度和一致性表明长期被视为备选方法的 de novo 测序现在可以与数据库搜索并列为一个严肃的、在某些情况下更优的选项。
宏观视角:迈向完整且无偏的蛋白读取
对非专业读者来说,关键结论是 DiNovo 使人们更容易在不受限于参考数据库的情况下准确读取蛋白片段。通过将被良好支持的序列信息翻倍或三倍并提供内置的错误检查,这一方法为发现不熟悉的蛋白、追踪微妙变异以及探索成分仍多未知的复杂混合物打开了大门。简而言之,通过将镜像酶与深度学习和严谨的统计学相结合,DiNovo 有助于将嘈杂的谱图痕迹转化为更清晰、更可信的蛋白质图景,这些蛋白质构成了健康与疾病的基础。
引用: Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
关键词: 蛋白质组学, de novo 肽测序, 质谱, 深度学习, 镜像蛋白酶