Clear Sky Science · zh
靶向新生嘧啶合成使PARP抑制剂耐药的卵巢癌对铜介导的ATR失活产生脆弱性
这项研究为何重要
许多卵巢癌患者接受的治疗是破坏肿瘤细胞修复受损DNA能力的药物。这类药物称为PARP抑制剂,起初常能取得良好效果,但肿瘤常会适应并复发。本研究揭示了一种携铜药物与一种关键代谢弱点如何可能将耐药的卵巢癌推向临界点,提示更聪明的联合疗法和更持久的治疗反应。
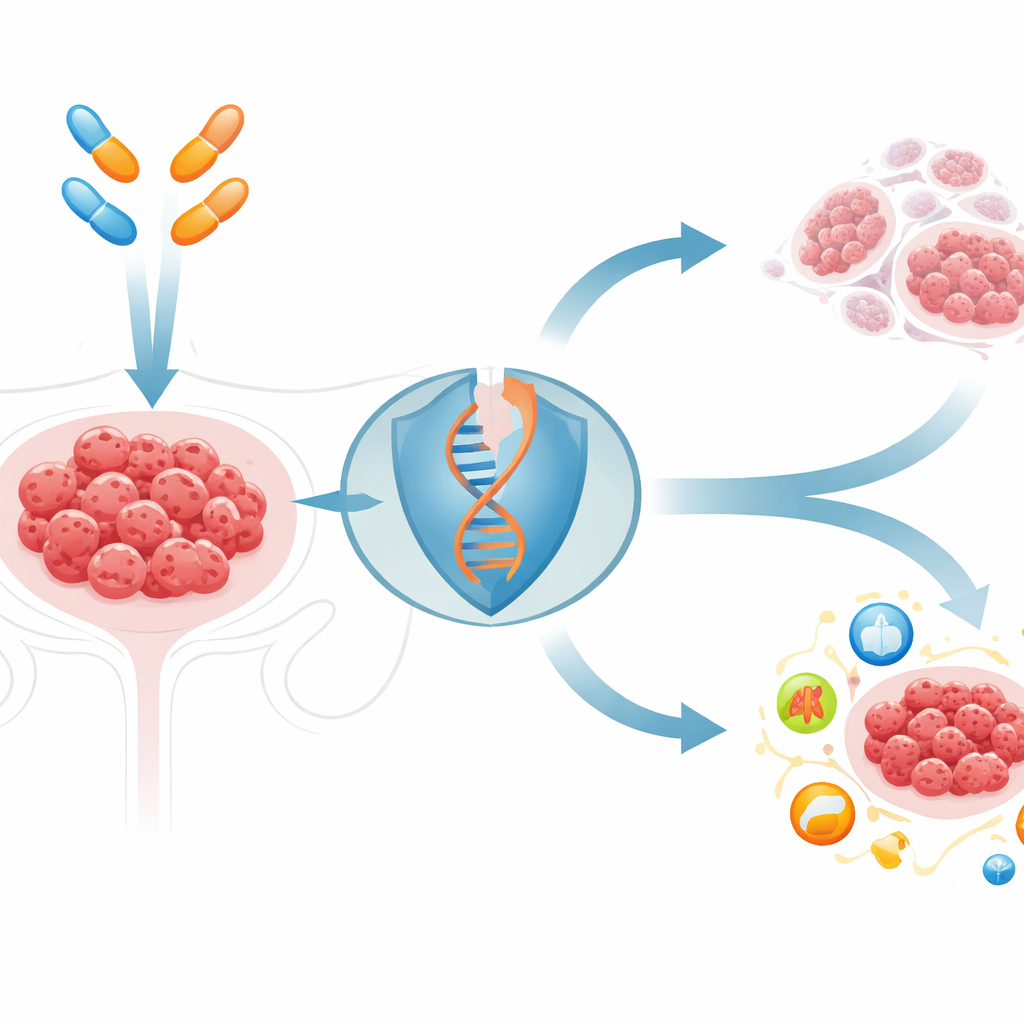
攻破顽固的肿瘤防线
PARP抑制剂利用一些癌细胞修复断裂DNA时的缺陷。它们在具有遗传性BRCA基因缺陷的肿瘤中效果最佳,但大多数卵巢癌BRCA完整,对此类药物反应差或仅短暂有效。研究人员在标准PARP抑制剂的基础上筛选了144种与细胞死亡相关的化合物,发现一种名为elesclomol的药物尤为突出。elesclomol将铜运入细胞。在BRCA正常的卵巢癌细胞和小鼠肿瘤中,与PARP抑制剂联合使用时,这种增加细胞内铜的药物显著加剧了DNA损伤并使肿瘤缩小,远胜于任一药物单用,且在健康器官中未见明显毒性。
铜干扰DNA修复开关
为了解为什么铜会让PARP阻断更致死,研究团队考察了以ATR蛋白为中心的主要DNA损伤信号通路。该通路帮助细胞在DNA复制受压时存活——正是PARP抑制剂造成的情形。在那些经初始药物暴露后幸存的肿瘤细胞中,ATR及其配对蛋白CHK1被强烈激活,而相关通路(ATM‑CHK2)则保持沉默。详细的生化检测和计算机引导的结构建模显示,铜直接结合到ATR的辅助蛋白ATRIP的特定半胱氨酸位点。这种结合改变了ATRIP的构象,使其与ATR的接触被破坏,从而关闭了ATR‑CHK1信号,导致损伤的DNA无法修复,使接受PARP治疗的癌细胞更易死亡。
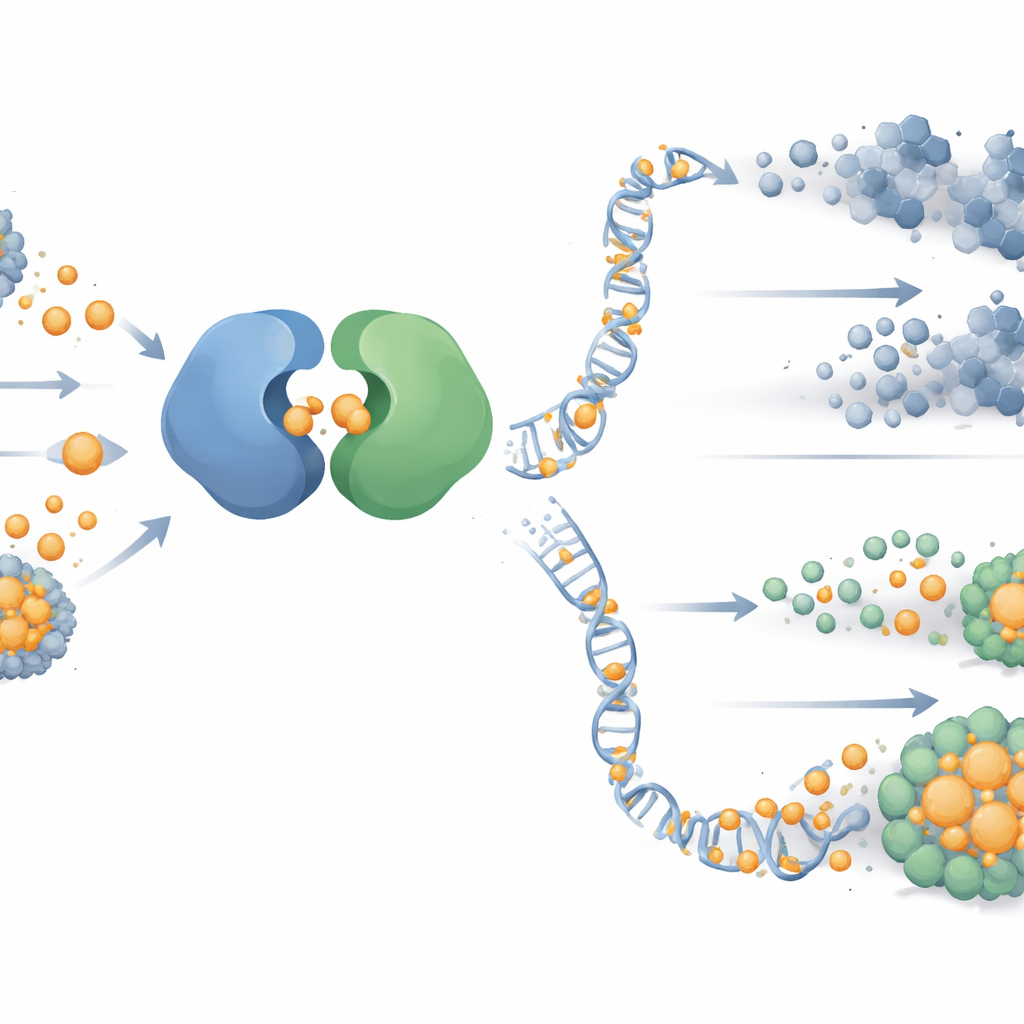
核苷酸燃料的隐秘作用
即便ATR和PARP同时受损,一些癌细胞和残余肿瘤仍能存活。为弄清原因,研究者对药物适应性细胞内数百种小分子进行了描绘。他们发现一种被称为嘧啶的DNA构件,尤其是通过“新生”(de novo)途径合成的嘧啶,其水平显著上升。示踪实验证实,耐药细胞将来自谷氨酰胺的更多氮用于合成新的嘧啶,而嘌呤类构件并未出现类似增加。向培养物中补充尿苷或胸苷等额外嘧啶成分,会削弱PARP加ATR或基于铜治疗的杀伤效果,提示充足的DNA构建供给可帮助肿瘤耐受原本致命的DNA损伤。
打击代谢弱点
研究团队接着测试阻断此嘧啶供应线是否能堵住肿瘤的逃生口。他们使用了BAY‑2402234,这是一种抑制新生嘧啶合成关键酶DHODH的试验性药物。在卵巢癌细胞系和患者来源的类器官中,加入DHODH抑制剂使对PARP加ATR或铜治疗已产生耐受性的细胞恢复敏感,清除先前耐药的细胞。在小鼠肿瘤和八个患者来源的异种移植模型中,那些对单用PARP——甚至对PARP联合ATR或铜阻断——产生耐药的肿瘤,在同时阻断嘧啶合成时出现强烈的生长抑制。天然嘧啶代谢物水平较高的肿瘤对基于PARP的治疗最难以应对,但在靶向该代谢通路后出现了响应。
这对患者可能意味着什么
本研究揭示了PARP抑制剂耐药的卵巢癌中两个相互关联的脆弱点。首先,铜可被用作精准工具,通过撬开ATR与ATRIP的结合来使关键DNA修复开关ATR失活,从而增强标准DNA靶向药物的效力。其次,通过提高嘧啶生成而产生适应性的肿瘤,会变得依赖该代谢通路,阻断它可以使肿瘤重新对治疗敏感。实际上,这些发现支持定制的联合治疗策略:对于嘧啶依赖较低的肿瘤,可考虑PARP抑制剂加ATR靶向药;而对于在代谢上易产生耐药的肿瘤,可采用同时阻断嘧啶合成的三联方案。尽管仍需进一步临床验证,这项工作为克服卵巢癌中最顽固的药物耐受形式绘制了更清晰的路线图。
引用: Nan, Y., Wang, K., Hu, M. et al. Targeting de novo pyrimidine synthesis confers vulnerability to copper-mediated ATR inactivation in PARP inhibitor-resistant ovarian cancer. Nat Commun 17, 3142 (2026). https://doi.org/10.1038/s41467-026-70001-5
关键词: 卵巢癌, PARP抑制剂, 铜疗法, DNA修复, 嘧啶代谢