Clear Sky Science · zh
在亚介观蛋白复合体中高效采样大尺度构象通路和中间构象
观察蛋白质的运动
许多维持生命的分子并不像刚性的乐高积木,而更像不断改变形状的微型机器。这些运动驱动了产生能量、修复DNA以及病毒进入细胞等过程。像冷冻电子显微镜这样的实验可以捕捉到这些形态的某些瞬间,但无法记录那些转瞬即逝的中间步骤。本文介绍了eBDIMS2,一种新的计算方法,能够为蛋白质运动“补上缺失的帧”,即使对于先前过于庞大和复杂、在普通计算机上无法模拟的巨型分子机器也适用。
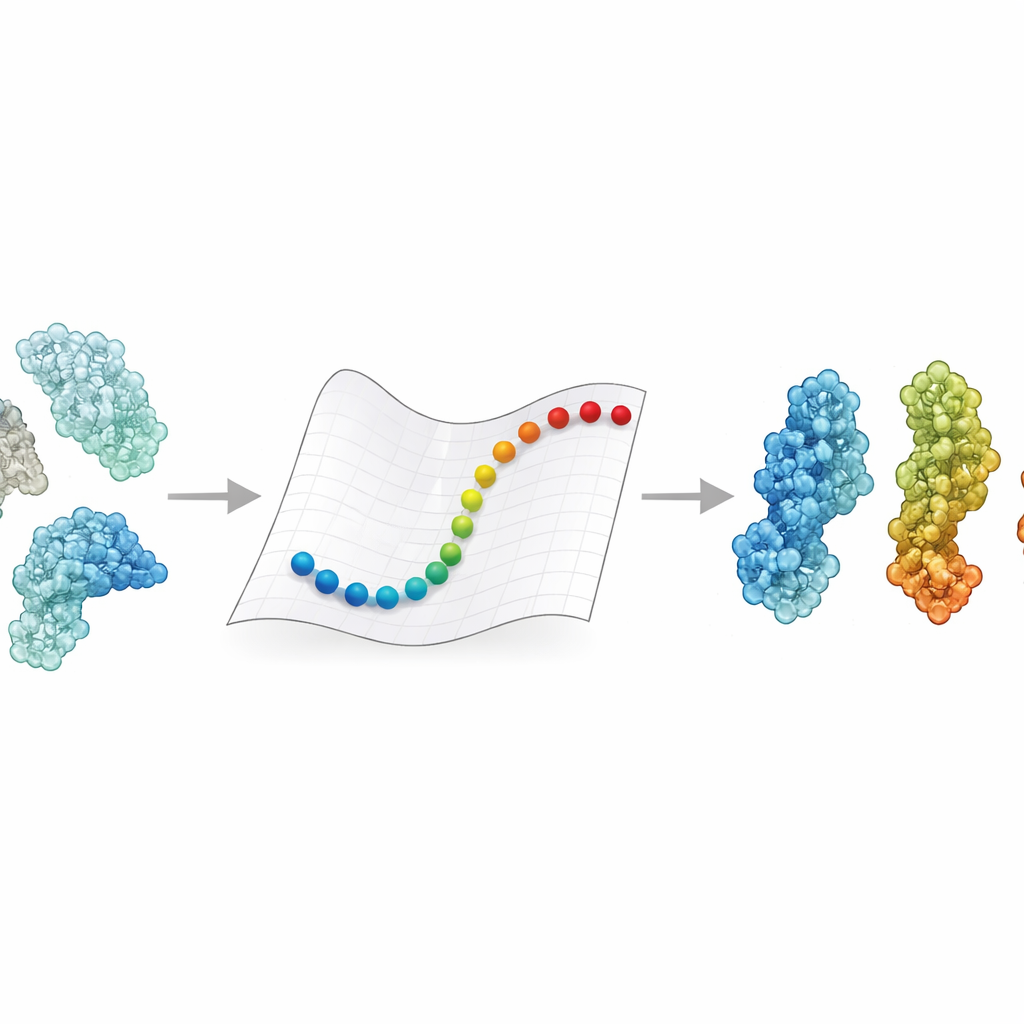
为什么蛋白质构象变化很重要
蛋白质很少保持单一姿态。它们会根据电压变化、pH或配体结合等信号开合、扭转或弯曲。这些变化可能决定酶是开启还是关闭,或受体是捕获病毒还是让其逃逸。实验为我们提供了几个关键构象的详细快照,分子动力学模拟理论上可以通过随时间追踪每个原子来连接这些快照。但对目前冷冻电镜所见的巨大复合体——常常重达数十万到数百万道尔顿——进行这样的追踪,通常需要超级计算机和数周的计算。因此,对于许多医学上重要的巨型装置,我们仍然不知道一种状态如何转变为另一种状态。
穿越蛋白质构象景观的更快路径
eBDIMS2 通过简化蛋白质的表示和运动计算方式来走捷径。它不是跟踪每个原子,而是将每个氨基酸看作一个由弹簧连接的点,构成弹性网络。这些弹簧捕捉了蛋白质不同部分趋于一起运动的倾向。该方法随后使用布朗动力学——模拟在液体中热扰动的数学规则——将结构从一个已知的实验态推动到另一个。关键在于,eBDIMS2 仅关注真正重要的相互作用,使用距离阈值和并行计算来降低计算成本。这使程序的扩展性从大致二次增长接近线性增长。在实践中,这意味着近两百万道尔顿规模的巨大组装体的转变可以在桌面计算机上几小时内探索完成,而不再是基本无法触及的任务。
将路径与真实蛋白对照
为了检验这些快速路径是否具有生物学意义,作者组装了包含47种大型蛋白质和另外15个复合体的集合,总计数百个结构,主要由冷冻电镜解析。他们使用主成分分析(一种挑出蛋白质主要运动方式的统计工具)将这些结构组织成构象景观,例如开放、闭合、活性或非活性等。随后让 eBDIMS2 在该景观中连接成对的端态。得到的路径被投影回相同的低维图谱,以判断它们是否沿着平滑路线经过实验观测到的中间体。超过30%的体系中,模拟路径与未被提供为输入的中间构象在几埃范围内接近。对于挑战性较高的例子,如DNA修复酶DNA-PKcs或冠状病毒刺突蛋白,这些粗粒化路径也与代价更高的原子级模拟(包括定向分子动力学和先进增强采样运行)高度重叠。
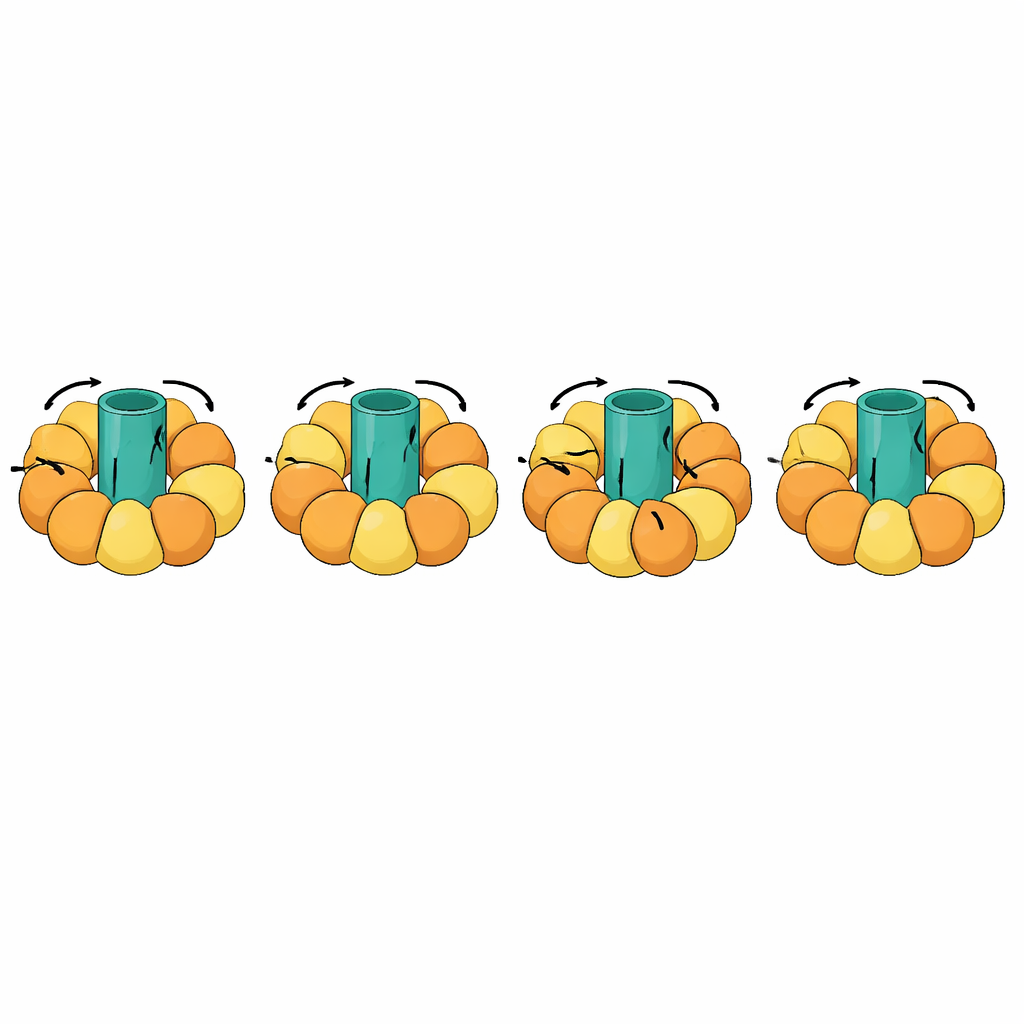
追踪巨型分子机器
其中一个最引人注目的测试涉及旋转类机器,例如ATP合酶,它通过使膜内的旋转转子耦合周围亚基的开合运动来生成细胞的能量货币。这些转变异常复杂:分子的一部分必须保持刚性并作为整体旋转,而其他部分则在有节奏的周期中弯曲。eBDIMS2 为这样的准刚体片段和存在缺失段的非完整实验模型(在冷冻电镜中常见)引入了特殊处理。有了这些特性,它可以模拟ATP合酶的完整旋转周期以及其他大型复合体如分子伴侣、受体和病毒组装体。在整个过程中,生成的中间构象避免了一些竞争方法产生的严重畸变,并且可以被整理成适用于药物设计计算或更长、更精细模拟的原子级模型。
这对生物学和医学意味着什么
研究表明,eBDIMS2 能可靠地勾勒出对传统模拟而言难以触及的体系中已知蛋白构象之间的主要通路。它并不替代详尽的原子级动态电影,也不提供精确的能量和时间信息,但它提供了一种快速且物理基础的方式来描绘大型分子机器可能的运动路径,仅需一对实验结构作为输入。随着结构数据库中越来越多与癌症、感染及其他疾病相关的大型蛋白装配体的多种状态被记录,这一方法为研究人员提供了一个便捷工具,用于连接这些状态、提出合理的中间构象,并指导在哪里用更高分辨率的方法或有针对性的药物设计进行后续探索。
引用: Scaramozzino, D., Lee, B.H. & Orellana, L. Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes. Nat Commun 17, 2202 (2026). https://doi.org/10.1038/s41467-026-69809-y
关键词: 蛋白质动力学, 分子模拟, 冷冻电子显微镜, 构象通路, 粗粒化建模