Clear Sky Science · zh
使用第14族烯丙基阿特兰烷在α‑氧烯酮的烯丙化反应中实现非螯合控制
以更高精度构建分子形状
许多药物和天然产物只有在原子以恰当的三维排列时才发挥作用。因此化学家投入大量精力学习如何从分子的一侧或另一侧“推进”新片段。本文介绍了一种将烯丙基——短的三碳链——连接到一类常见分子上的新方法,从而获得以往很难合成的镜像结构。
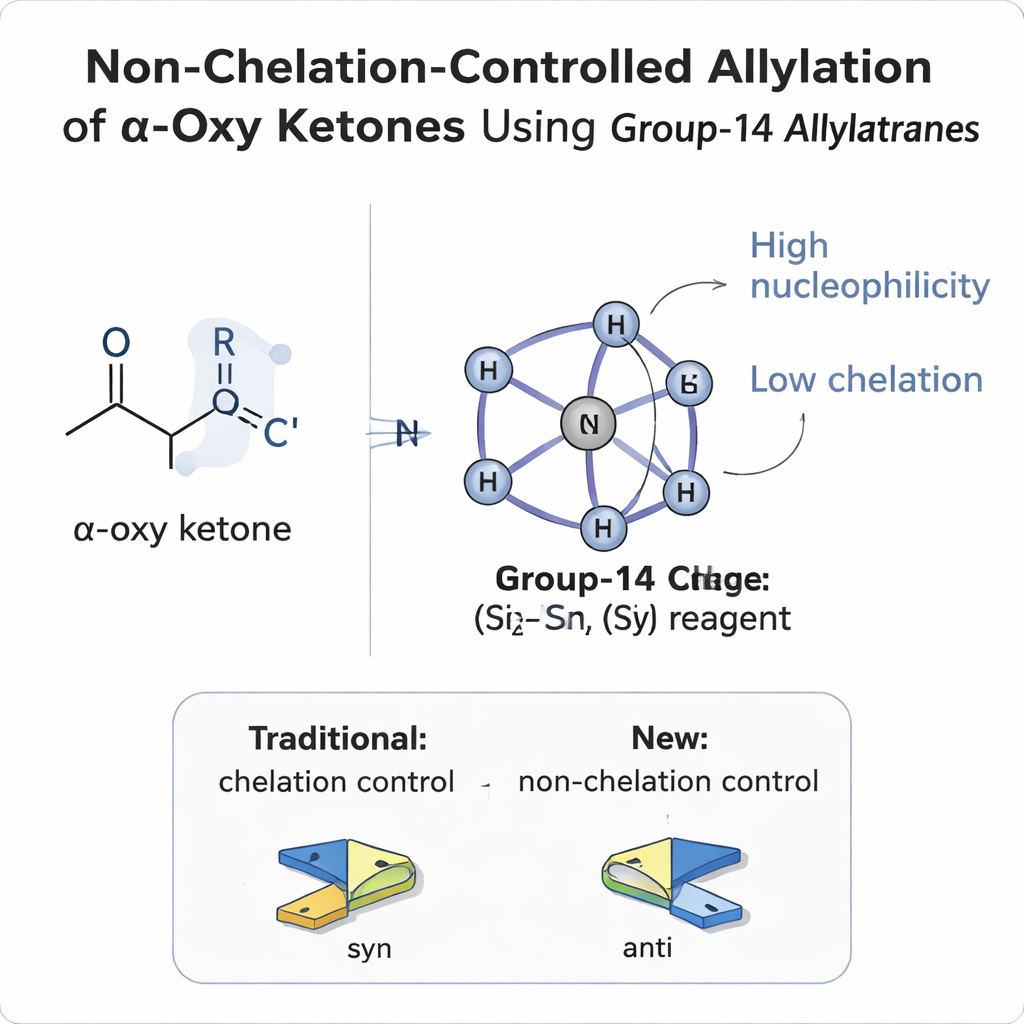
为什么控制分子一侧如此困难
当一个新基团加到平面的碳‑氧双键(羰基)时,它可以从任一面进攻,就像球从上方或下方击打硬币一样。如果在相邻的碳(α‑位)上已有取代基,产物会有两种不同的三维构型,称为仲兄弟异构体。数十年来,化学家依赖简单模型——Felkin–Anh、极性Felkin–Anh、Cram和螯合模型——来预测试剂偏好进攻的一侧。在α‑氧羰基化合物中,该邻位取代基是含氧基(如醚或酯),氧原子通常像爪子一样抓住金属试剂。这种“螯合”把分子锁定在一种构象,几乎总是导致所谓的顺式产物,其中新形成的醇基位于碳链的同一侧。
长期存在的氧邻位问题
虽然螯合通路非常有用,但也有局限:它强烈偏向顺式产物,使获得相反的反式构型——两醇基指向相对两侧——变得困难。对于α‑氧醛(羰基碳上至少连有一个氢)而言,利用特殊硅试剂和谨慎选择的路易斯酸已经能实现少量反式产物。但对于活性较低且在复杂目标中更常见的α‑氧酮,挑战更大。更强的亲核试剂往往具有更高的路易斯酸性,这反过来又助长了化学家希望避免的螯合。因此,在“足够活泼”和“不与氧太强结合”之间取得平衡一直是一个未解决的中心问题。
一类打破规则的笼状试剂
作者引入了一类称为烯丙基阿特兰烷的新型试剂,围绕第14族元素——硅、锗和锡——构建于刚性笼状框架中。在这些分子里,内部的氮原子跨越笼体与中心原子成键,形成高度配位、几乎被包裹的金属中心。这种设计有两个关键效果。其一是增强了所连烯丙基的亲核性,使其更愿意形成新的碳‑碳键;其二是抑制了中心原子的路易斯酸性,从而不太倾向于与底物的氧原子强烈结合。量子化学计算和核磁共振数据证实电荷局限于烯丙基片段,而与硅中心的电子通讯被减弱,这解释了该试剂为何既强劲又不易发生螯合。
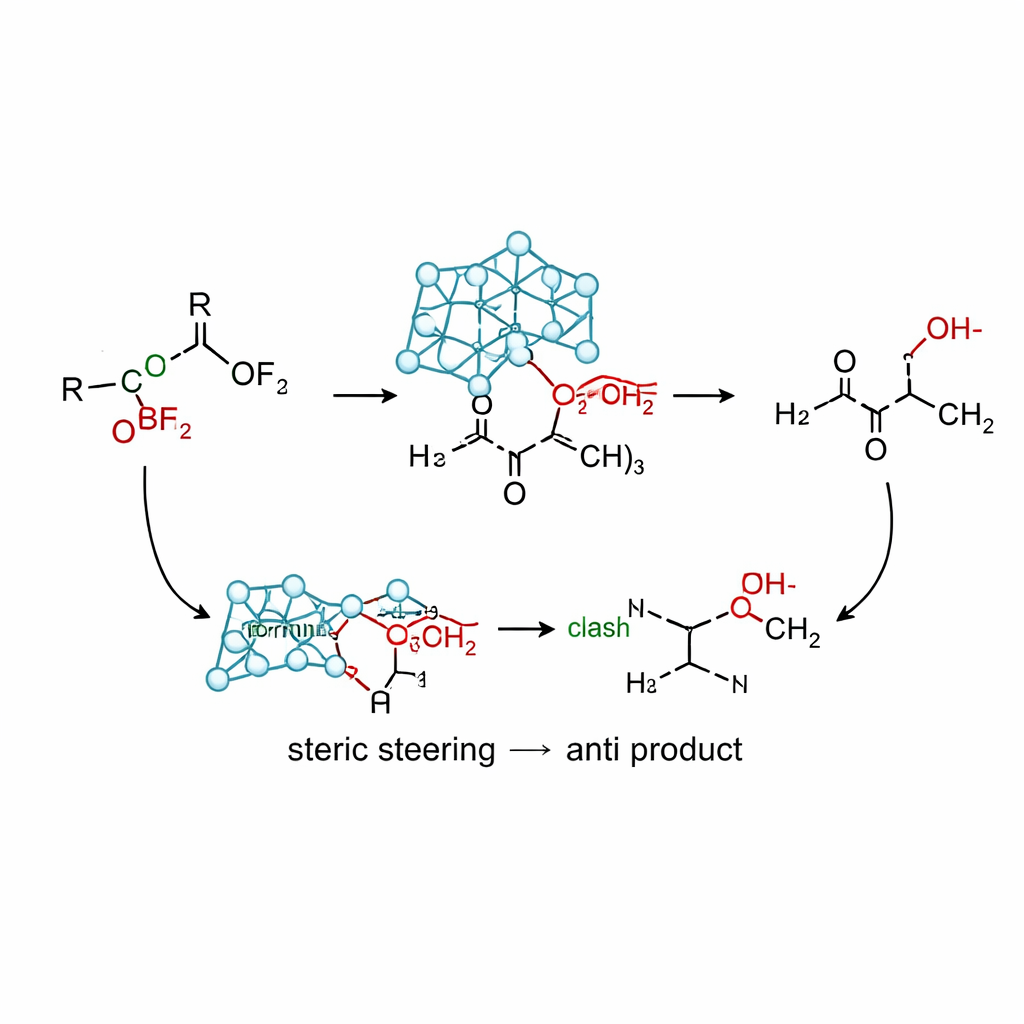
新反应在实践中的工作原理
使用该试剂的硅版本——烯丙基硅阿特兰烷,配合温和的路易斯酸(三氟化硼),团队建立了能从各种α‑氧酮高产得到反式高同族醇的条件。基准实验表明,许多传统的烯丙基来源——基于锡、铟、镁、锂和简单硅烷的试剂——要么偏向顺式产物、要么产物混合、要么发生分解。相比之下,烯丙基硅阿特兰烷在带有甲氧基、异丙氧基、苯氧基、乙酰氧基、硅醇基和氨氧基的底物以及各种芳环和环酮上,常常实现超过95:5的反/顺选择性。计算结果表明存在一种非螯合路径,其中体积大的阿特兰笼体引导烯丙基的进攻趋向类似Cram模型的构象:氧取代基位于羰基的对侧,而笼体与邻近苯环之间的位阻排斥促使进攻轨迹走向产生反式产物的一侧。
对药物与天然产物合成的影响
该方法对更为刚性的环状体系也有效:烯丙基硅阿特兰烷的体积促使从更不受阻的位置进攻;用于转移取代烯丙基的专用衍生物也能实现同样高的反式选择性。由于所得含有高同族片段的反式1,2‑二醇是生物活性分子(包括酶调节剂候选物)中的常见基元,这一转化为化学家提供了一条可靠途径,以获得先前需要多步变换或实际不可行的结构变体。重要的是,通过在可螯合的锡体系和新的非螯合硅阿特兰体系之间切换,同一底物可以被定向生成顺式或反式产物,从而实现对分子形状的精确控制。
简单说,这意味着什么
本质上,研究者构建了一个智能的烯丙基递送工具,能从“非优选”一侧打击目标碳,同时不被邻近氧原子束缚。通过在硅周围精心设计一个笼状结构,他们将活性与黏性解耦:试剂既足够强以构建新键,又不至于因抓住周围原子而破坏期望构型。对非专业读者而言,这意味着化学家现在对塑造支撑许多药物与天然产物的三维分子结构有了更好的掌控,扩展了未来药物与复杂合成分子的设计空间。
引用: Tsutsui, Y., Shiga, K., Konishi, A. et al. Non-chelation control in allylations of α-oxy ketones using group-14 allylatranes. Nat Commun 17, 2019 (2026). https://doi.org/10.1038/s41467-026-69732-2
关键词: 立体选择性烯丙化, α‑氧酮, 烯丙基硅阿特兰烷, 非螯合控制, 高同族醇