Clear Sky Science · zh
XL-MSDigger:一种基于深度学习的、多用途交联质谱解决方案
看清蛋白如何组合在一起
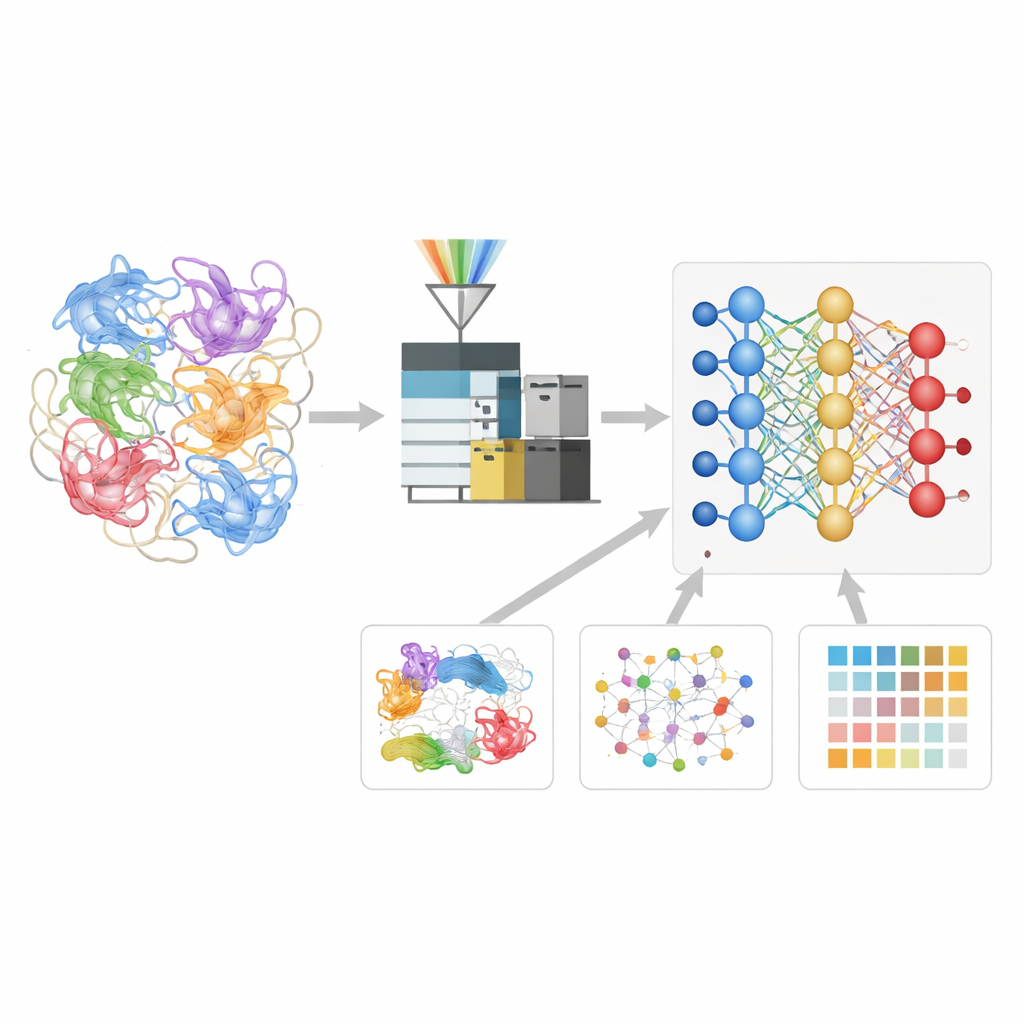
我们体内的每一个过程不仅依赖于蛋白质折叠成正确的形状,还依赖于它们找到合适的伙伴。然而,实时观测这些分子关系向来困难重重。这项研究介绍了 XL-MSDigger,一款软件平台,利用现代人工智能从一种噪声很大的实验技术——交联质谱中提取更清晰的信号,帮助科学家绘制细胞内蛋白质的排列方式及其相互作用网络。
解开拥挤的分子世界
为了研究蛋白质的构成与连接方式,研究者常使用交联质谱。在这种方法中,小分子“桥梁”会连接彼此接近的蛋白片段。被连接的片段随后被切割成肽段,并在质谱仪中测量其质量。原则上,碎片的模式可以揭示哪些蛋白片段在空间上靠近,就像找到哪些书页被夹在一起一样。但在实际操作中,所得数据极其复杂。现有的计算工具大多只考察基本的质量信息,难以应对海量可能组合,导致漏检和误配现象频繁。
教神经网络理解蛋白碎片的“语言”
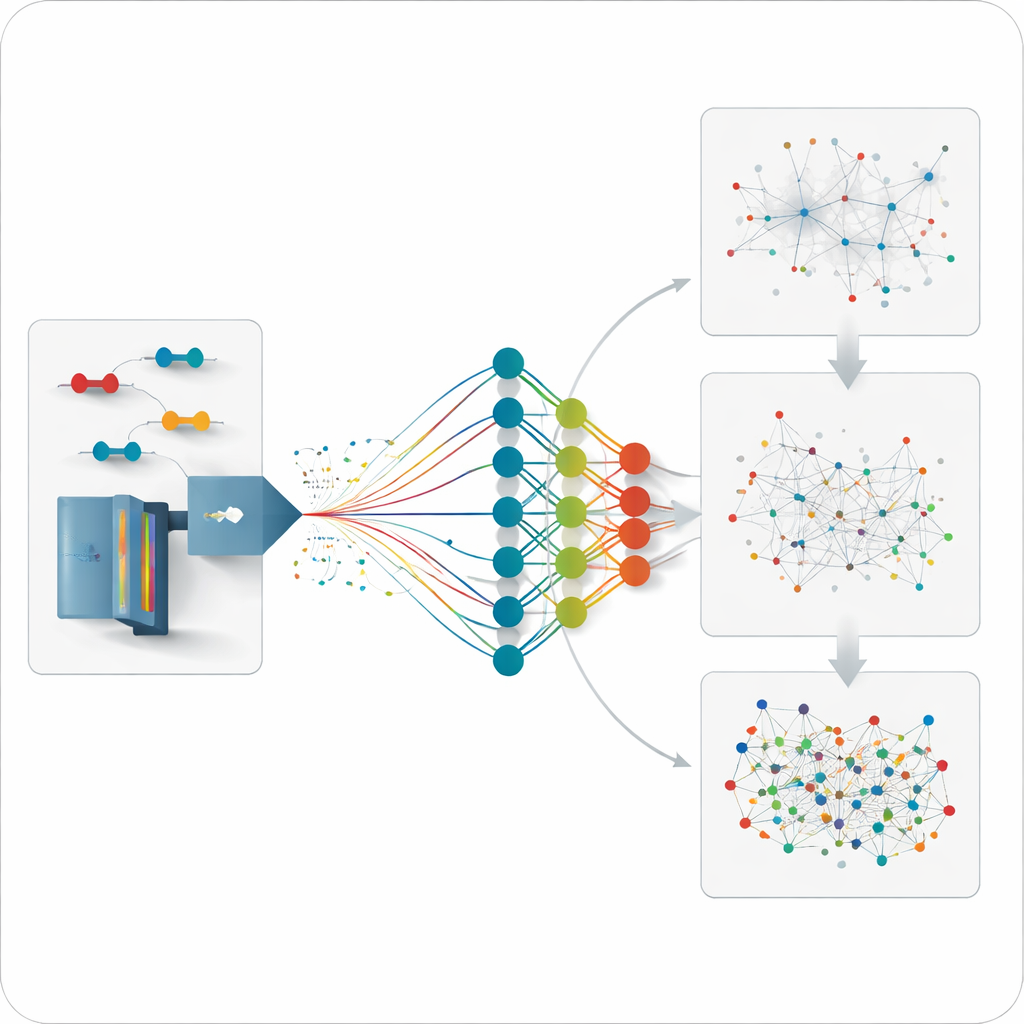
作者构建了一个名为 Deep4D-XL 的深度学习模型以更好地解释这些交联实验。他们首先通过对人类细胞的蛋白进行交联、将其裂解为肽段,并记录不仅是质量,还包括肽段在仪器中通过的保留时间以及在离子迁移室中的移动行为,从而建立了大规模参考集。每一对交联肽段都被编码输入模型,模型采用双胞胎式的“孪生(Siamese)”结构来分别读取两个肽段,并通过交叉注意力步骤将它们的信息融合。由此,网络学会预测任意新的交联肽段的三个关键属性:它应当何时在实验中出现、如何移动以及其碎裂图谱应是什么样子。
把预测变成更清晰的信号
XL-MSDigger 将这一预测引擎封装进针对两种主要数据采集方式的分析工作流程中。在传统的、目标导向的采集模式下,仪器会选择性记录其实时挑选出的离子的碎片。XL-MSDigger 对现有检索软件给出的初始匹配结果进行重评估,利用模型对每个候选肽段的预测行为作为参考。第二个神经网络在多个维度上比较预测与实验并赋予改进后的评分。该重评分步骤几乎将酵母和人样品中不同蛋白之间可置信检测到的交联数量翻倍,同时保持较低的错误率,从而揭示出远多于以往的蛋白—蛋白相互作用。
理解海量无偏数据的含义
一种更新的运行方式称为数据无关采集(DIA),它记录样品中几乎所有成分的碎片,虽然覆盖面更广,但会产生令人难以承受的数据量。到目前为止,尚无良好方法来估计这些得到的交联中有多少是真实的。XL-MSDigger 利用 Deep4D-XL 构建一个精心匹配的“诱饵”库(decoy),然后同时分析真实与诱饵条目。通过观察诱饵被误识别的频率,软件能够估算假发现率,并训练另一个神经网络将真信号与假信号区分开来。该重评分使得可置信的交联信号数量大约提升五倍,并在真实与诱饵模式之间产生清晰分离。
预测尚未被测量的信号
由于模型可以预测任何合理的交联肽段应当如何表现,团队进一步分析了从未被直接测量过的连接。他们生成中等规模的预测库,聚焦于选定的蛋白或相互作用网络,然后将无偏数据与这些库进行比对。这一策略发现了更多单体蛋白内部及重要伴侣蛋白之间的连接,其测得距离与已知三维结构吻合良好。它也恢复了传统、较有限实验库中遗漏的相互作用,尤其是那些丰度较低的连接。
为蛋白伙伴关系打开更清晰的窗口
对于非专业读者,关键结论是:XL-MSDigger 像一个高度训练的模式识别器,叠加在已然强大的实验方法之上。通过在多个维度上学习真实交联信号应有的样子,它能够在海量、混乱的数据集中筛选、剔除可能的伪信号,并挽救先前被隐藏的真实蛋白连接。尽管对整个蛋白组的全面应用仍需要大量计算资源,这项工作展示了将交联实验与深度学习结合能大幅提升我们对细胞内蛋白如何排列及相互会面的认识。
引用: Chen, M., Hao, Y., Huang, X. et al. XL-MSDigger: a deep learning-based, versatile solution for cross-linking mass spectrometry. Nat Commun 17, 2554 (2026). https://doi.org/10.1038/s41467-026-69489-8
关键词: 蛋白质相互作用, 交联质谱, 深度学习, 蛋白质组学, 数据无关采集