Clear Sky Science · zh
锌缺乏时KAT7依赖的组蛋白H3K14乙酰化受损的病理生理学意义
为何微量营养素对我们的健康至关重要
锌是一种微量金属,人体只需摄入极少量,却在无数蛋白质中发挥关键作用,维持细胞功能。当锌不足——无论因饮食、疾病或衰老——都与从生长不良到免疫力下降乃至脂肪肝等问题相关。本研究提出了更深层的问题:细胞如何感知锌短缺?这种短缺如何被转化为对基因活动和器官健康的持久改变?
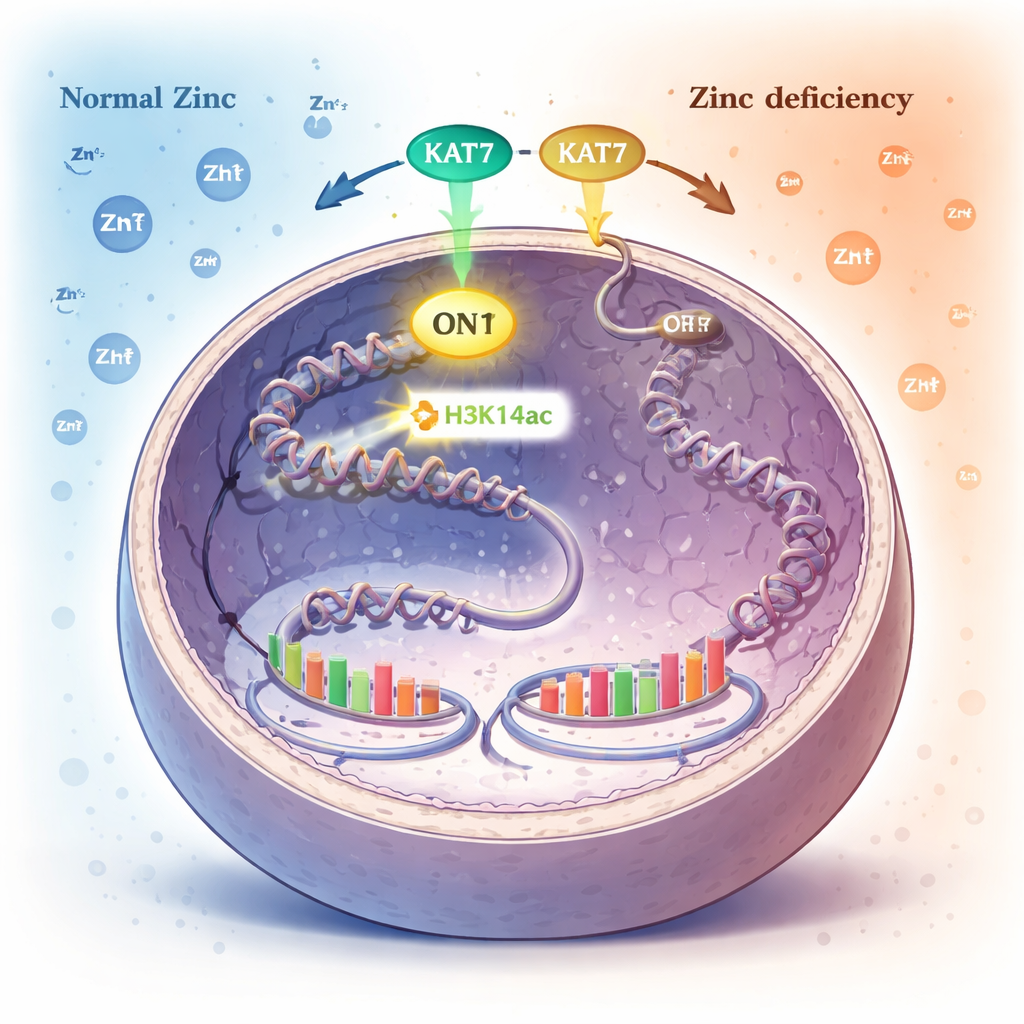
作为细胞内锌警报的DNA包装化学标记
在细胞核内,DNA缠绕在称为组蛋白的蛋白线轴上。细胞通过在这些组蛋白上添加或去除微小的化学标记来控制哪些基因被激活。其中一种标记是在组蛋白H3的特定位点上的乙酰化(H3K14ac),由名为KAT7的酶添加。作者发现,当锌变得稀缺时,这一H3K14ac标记的水平显著下降,而许多其他常见的组蛋白标记保持不变。这提示H3K14ac及其生成酶KAT7是感知细胞锌状态的关键标志。
锌如何维持关键酶的开启状态
通过系统性地使不同酶失活,研究人员证明KAT7是人类细胞中H3K14ac的主要来源。KAT7在其活性中心包含一个小的锌配位结构。当细胞被置于锌缺乏状态时,KAT7在位点上放置H3K14ac标记的能力下降,尽管该蛋白仍位于细胞核并与其辅助伴侣保持结合。对纯化的KAT7片段的详细测试表明,该区域中正确结合的锌对其活性至关重要;破坏锌结合会使酶失活,而谨慎补充锌则可恢复其功能。本质上,KAT7表现出类似锌依赖的开关特性,控制特定的组蛋白标记。
将锌缺失转化为提升锌水平的基因变化
这种组蛋白标记的丧失究竟会导致什么?利用全基因组定位,团队发现H3K14ac特别富集于增强子区域——对附近基因进行精细调控的DNA片段。在锌缺乏条件下,许多增强子处的H3K14ac被去除,且标记丧失越多,邻近基因活性的变化越强烈。一个突出基因是ZIP10,它编码位于细胞膜上的锌输入蛋白。当ZIP10的增强子处H3K14ac下降时,ZIP10在膜上的表达上升,使更多锌进入细胞。阻断KAT7或阻止H3K14ac丧失会干扰这一反应,减少锌摄取,即使在补充锌后也是如此。这表明细胞将锌匮乏转化为一种表观遗传信号,增强锌输入机器以恢复平衡。
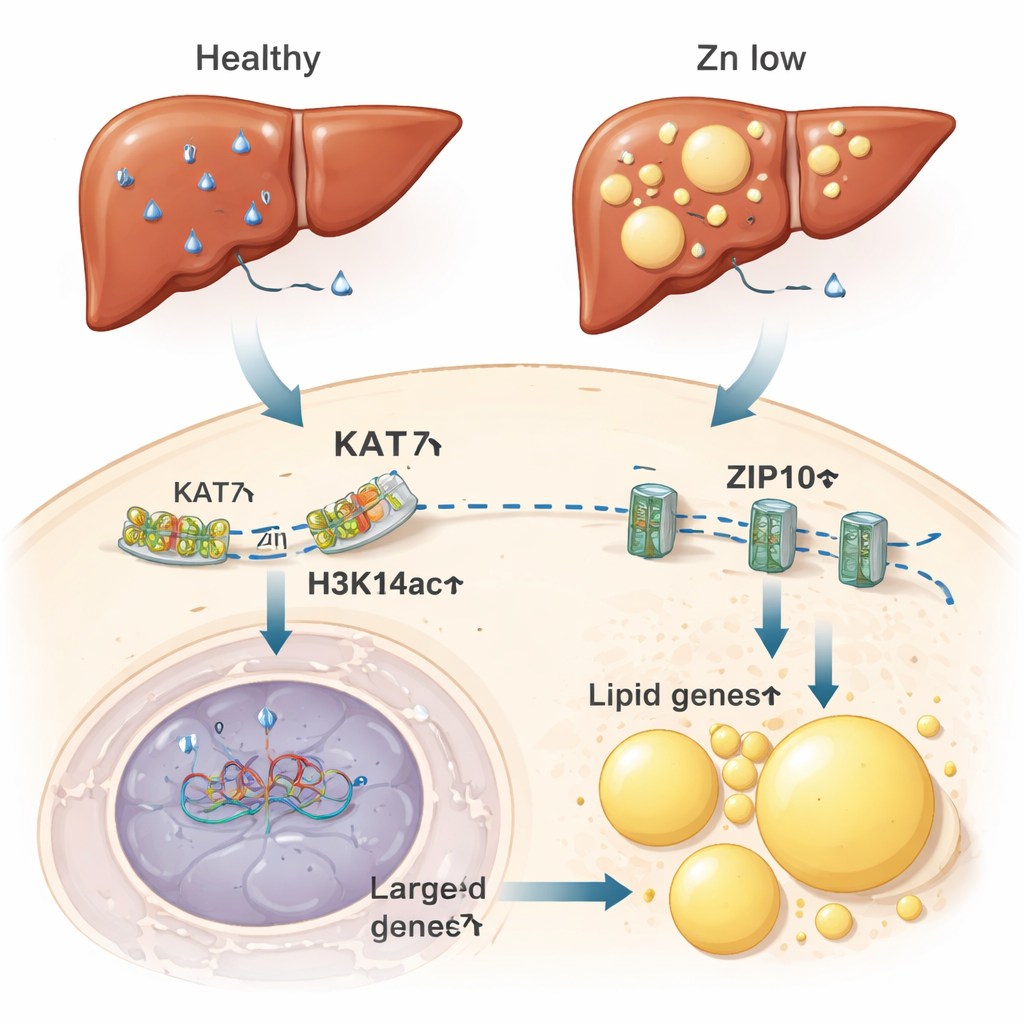
从缺锌细胞到脂肪肝的演变
作者接着探讨这一对锌敏感的开关在整只动物体内是否有后果。在给予缺锌饮食的小鼠中,作为锌和脂质代谢中心的肝脏表现出锌水平下降、H3K14ac降低以及KAT7活性减弱。这些变化伴随着促使脂肪储存和脂滴形成的基因表达增加。缺锌小鼠的肝脏脂肪堆积程度可与高脂饮食小鼠相媲美。值得注意的是,仅用药物降低KAT7活性,即使不改变饮食中的锌,也足以促进肝细胞的脂肪堆积。相反,补充额外锌可减轻高脂饮食导致的脂肪积累。
这对人类疾病风险意味着什么
将发现置于临床背景中,研究人员回顾了测量人类肝组织中锌水平的研究。多项报告显示,患有脂肪肝及相关疾病的人群肝脏锌含量显著低于健康对照组。结合小鼠实验证据,这提示慢性锌缺乏可能通过使KAT7失活、抹去H3K14ac标记并持续上调有利于脂肪储存的基因,从而促进脂肪肝的发生。简言之,这项工作揭示了一个内部的“锌‑到‑表观遗传学”回路:当锌下降时,一种锌依赖的酶失去活性,改变DNA的包装方式,这一改变短期内有助于细胞增加锌摄入,但长期则可能推动肝脏走向不健康的脂肪积累。
引用: Fujisawa, T., Takenaka, S., Maekawa, L. et al. Pathophysiological significance of impaired KAT7-dependent histone H3K14 acetylation during zinc deficiency. Nat Commun 17, 1710 (2026). https://doi.org/10.1038/s41467-026-69476-z
关键词: 锌缺乏, 表观遗传学, 肝脏脂肪, 组蛋白乙酰化, 锌转运蛋白