Clear Sky Science · zh
CAPN1 激活因子 CD99L2 的功能缺失变异导致 X 染色体连锁痉挛性共济失调
这对有不明原因运动问题的家庭意味着什么
许多人多年来一直受行走困难、肌肉僵硬或平衡与言语问题困扰,却始终查不出真正病因。本研究展示了现代 DNA 检测如何最终为其中一些家庭提供答案。研究者不仅比较了用于罕见运动障碍的不同基因检测方法,还发现了一种先前未知的 X 染色体连锁痉挛性共济失调的病因,这提示相关的生物通路可能在更常见的脑疾病中也具有重要作用。
在罕见病的干草堆中寻找遗传学的针
罕见运动障碍,如共济失调(动作不稳)和痉挛性截瘫(腿部僵硬无力),通常被怀疑具有遗传性,但对大多数患者来说,常规检测结果是阴性。研究小组在六年间随访了德国及欧洲各地被转诊怀疑罕见运动障碍的 2,811 名个体。首先,他们使用传统的靶向检测,搜索少数基因中的已知重复扩增,这类检测约能在 11% 的病例中给出答案。随后,他们采用外显子测序——只读取基因组中编码蛋白的部分——在大约 19% 的患者中找到明确的遗传学解释,尤其是在伴有痉挛性的患者中。
用全基因组测序超越标准检测
为了进一步推进,科学家们使用了全基因组测序,几乎读取了一个人全部 DNA,包括标准检测与外显子测序可能遗漏的区域。在 486 名接受更全面检测的个体中,诊断率约提高了 7.5 个百分点,这主要因为基因组测序更善于发现结构重排和重复扩增等复杂变异。研究还显示,仔细记录的临床信息——尤其是具体症状描述、较年轻的检测年龄,以及痉挛与其他运动问题并存——有助于预测谁更可能获得明确的遗传学诊断。
发现一种新的 X 染色体连锁痉挛性共济失调病因
即便经过这些广泛检测,仍有许多患者未能确诊。研究者汇集了超过 13,000 名个体的遗传数据,采用“基因负担”方法,比较患者与未受影响对照中哪些基因携带可疑变异的频率更高。这一分析不仅指向已知的致病基因,还强烈提示了一个先前被忽视的 X 染色体基因 CD99L2。通过整合来自欧洲多个家庭的结果,他们确定了来自 20 个家庭的 25 名受累男性携带该基因的破坏性变异。这些男性通常在中晚年出现行走问题、下肢僵硬、言语不清,有时并伴有平衡困难,而女性携带者大多无明显症状——这些表现符合 X 染色体连锁疾病的模式。所发现的变异主要导致正常蛋白功能丧失或关键结构缺失,强烈表明基因功能缺失可致病。
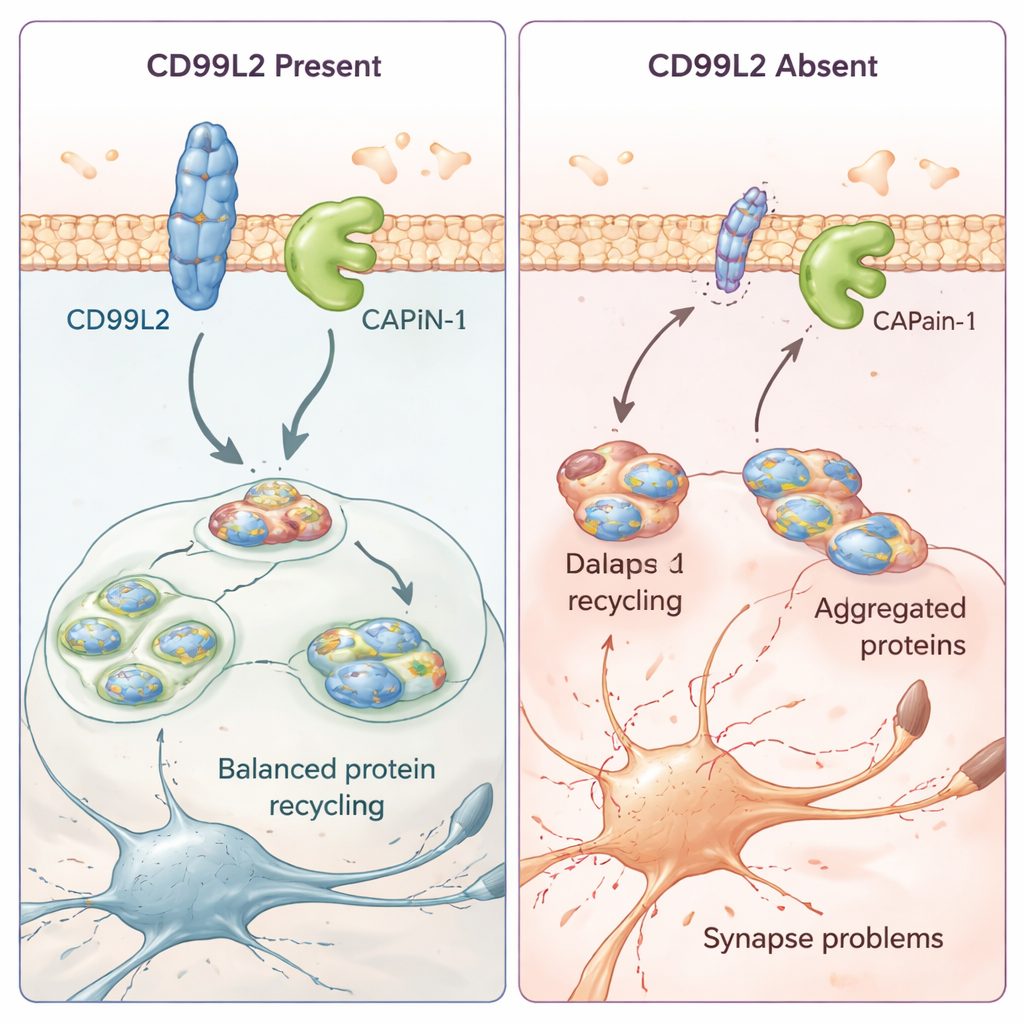
一个小型膜蛋白如何帮助保护脑细胞
为了弄清 CD99L2 在细胞中的实际功能,研究团队使用了细胞模型和患者来源的皮肤细胞。他们发现 CD99L2 蛋白位于细胞膜上,通常被带有小型“泛素”标签,这些标签控制其在被降解前存留的时间。CD99L2 能物理性结合钙激活的蛋白酶 calpain-1(CAPN1),该酶可切割其他蛋白并有助于维持突触——神经细胞之间接触点——的健康。当 CD99L2 存在且结构完整时,它有助于以受控方式开启与关闭 calpain-1,然后自身被切割并回收。当 CD99L2 缺失或结构改变时,calpain-1 的激活受到影响。在患者细胞中,这通常伴随许多与突触和神经细胞通讯相关基因的活动紊乱,提示细微但广泛的脑回路改变可能是逐步出现运动障碍的基础。
这对今天和未来的患者意味着什么
对于那些原因不明的痉挛性共济失调或痉挛性截瘫的家庭,这项工作带来了两方面的进展。首先,它表明早期采用全基因组测序并配合细致的临床描述,可以显著提高获得明确遗传诊断的概率。其次,它将 CD99L2 纳入控制 calpain 活性的基因名单,而这一通路已在其他罕见共济失调以及像阿尔茨海默病和帕金森病等常见病中被牵涉。通俗来说,这项研究揭示了一个帮助维持脑细胞平衡的“开-关”开关;当该开关损坏时,神经细胞会缓慢退化,导致僵硬和协调性下降。理解这个开关最终可能为调整 calpain 活性并保护各种神经疾病中的脑细胞的治疗方法打开大门。
引用: Menden, B., Incebacak Eltemur, R.D., Demidov, G. et al. Loss-of-function variants in the CAPN1 activator CD99L2 cause X-linked spastic ataxia. Nat Commun 17, 1698 (2026). https://doi.org/10.1038/s41467-026-69337-9
关键词: 痉挛性共济失调, 罕见运动障碍, 基因组测序, CD99L2, calpain-1