Clear Sky Science · zh
使用 AlphaFold 获得本征无序蛋白的原子分辨率构象集合
为何易变形的蛋白质重要
我们的细胞中充满了从不固定为单一刚性构象的蛋白质。这些“本征无序”蛋白更像柔软的面条而非整洁折叠的机器,但它们在从细胞信号传导到神经退行性疾病等过程中的作用至关重要。由于它们不断移动和弯曲,以原子级细节捕捉它们的全套构象极为困难,通常需要多年实验和大量计算。本文介绍了一种将人工智能与物理规律结合的新方法,可以更高效地绘制这些不安分分子的构象图谱。
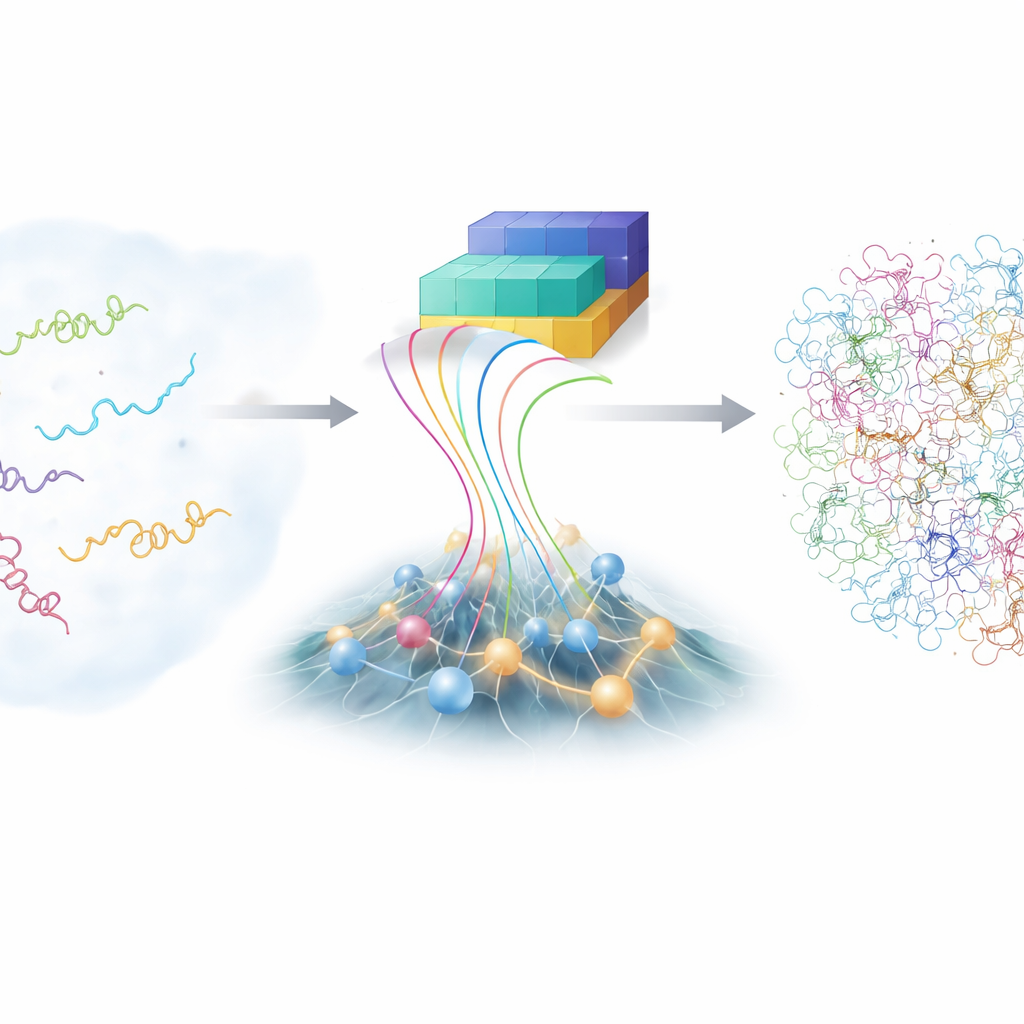
不安分分子的挑战
与教科书中的蛋白模型只显示一个整洁结构不同,本征无序蛋白(IDP)在可能构象的广阔景观中游走。这种灵活性有助于它们识别许多不同的伙伴,但也使得研究它们极为棘手。传统实验技术,如先进的核磁共振和 X 射线散射,可以报告跨越多种构象的平均性质,但无法给出每一种单独形式。原则上,原子级的计算模拟可以追踪 IDP 每个原子的运动,然而这些模拟代价极高且依赖精细调参的物理模型。因此,科学界只有有限的、准确且详尽的 IDP 构象集合可供学习。
将智能预测与物理规则结合
近年来,AlphaFold 系列深度学习工具通过从氨基酸序列预测蛋白结构而震惊了生物学界。然而对于无序蛋白,AlphaFold 的常规强项——猜测单一最佳构象——作用有限,因为 IDP 并非只有一个构象。AlphaFold 提供的有价值内容是关于链上不同片段相互接近或远离的概率信息。作者构建了一个名为 bAIes 的新框架,将这些来自 AI 的信息作为软引导,并与一个快速的基于物理的模型融合,该模型刻意从“随机线圈”视角出发,使链条在不偏好任何特定结构的情况下探索所有可能的弯曲和扭转。
从随机缠结到真实构象集合
首先,研究者构建了一个高效的物理模型,基于从数千个已知蛋白结构中提取的统计数据,重现完全无序蛋白链的行为。该模型作为“先验”——当我们一无所知时对 IDP 运动的基线预期。接着,bAIes 读取 AlphaFold 关于哪些残基对倾向接近的预测。它并不强行将蛋白推入单一模式,而是将这些提示转化为具有内置不确定性的温和距离约束,允许链仅在这些建议与更广泛物理图景一致时才去满足 AI 的指示。
与真实实验的比较验证
为了检验该方法的有效性,团队将 bAIes 应用于一组 21 个蛋白,范围从几乎完全的随机线圈到具有瞬时螺旋和多域的更复杂体系。对每个蛋白,他们将计算生成的构象集合与一系列探测局部细节及全局尺寸与形状的实验测量进行了比对。对于如与阿尔茨海默相关的肽 Aβ40 这样非常柔软的蛋白,简单的随机线圈模型已接近现实,bAIes 保持了这一良好一致性。对于部分结构化的蛋白,bAIes 通过正确捕捉短螺旋片段和紧凑片段出现与消失的位置,改善了与实验的匹配。关键在于,当 AlphaFold 过于自信并错误地预测出溶液实验显示为无序的稳定折叠时,bAIes 仍保持稳健,因为它明确允许对 AI 输入存在误差。
超越或匹配现有方法
作者随后将 bAIes 与在专用超算上运行的全原子模拟、将蛋白简化为珠粒的粗粒化模型以及基于模拟数据训练的新深度学习生成器进行了对比测试。在多项测试中,bAIes 在再现实验数据方面持续匹配或优于这些方法,同时计算开销远小于全尺度模拟。它还适用于超出简单 IDP 的体系,能够处理由若干刚性结构域通过柔性连接子连接的蛋白并恢复其溶液中的整体形状。当研究者进一步用实验数据微调 bAIes 构象集合时,一致性进一步提高,表明该方法可作为综合建模的强大出发点。
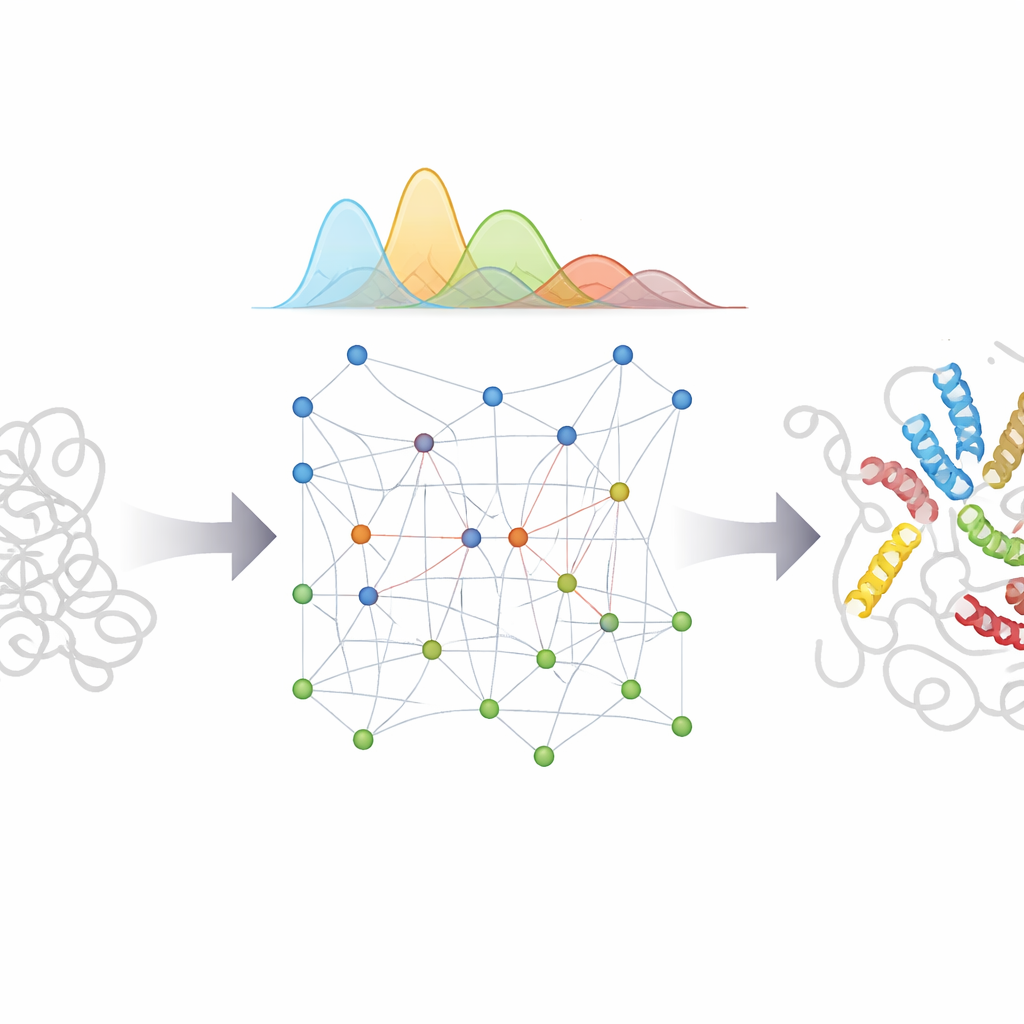
对生物学和医学的意义
通过将 AlphaFold 的模式识别能力与精心设计的物理模型及对不确定性的贝叶斯处理相结合,bAIes 为获得无序蛋白的详细“影片”而非单一快照提供了实用路径。这些具有原子细节的构象集合可以帮助科学家理解柔性区域如何识别伙伴、帕金森和阿尔茨海默等疾病中错误折叠与聚集如何起始,以及小分子如何与难以捉摸、不断变化的靶点结合。由于该方法高效且集成在开源软件中,它可被广泛采用以生成多种无序蛋白的现实构象集合,为实验提供指引并支持未来旨在预测不止一种结构、而是生命中最灵活分子所能采用的全部构象范围的 AI 系统。
引用: Schnapka, V., Morozova, T.I., Sen, S. et al. Atomic resolution ensembles of intrinsically disordered proteins with Alphafold. Nat Commun 17, 2399 (2026). https://doi.org/10.1038/s41467-026-69172-y
关键词: 本征无序蛋白, AlphaFold, 贝叶斯建模, 蛋白质构象集合, 结构生物学