Clear Sky Science · zh
使用分层可解释变换器可信地预测酶学编号
为何预测酶的“工作”至关重要
每个活细胞的运行依赖无数微小的化学机器——酶。每种酶都有特定的“工作”,这个工作通过酶学委员会(EC)编号来编码,这是一种类似邮政地址的四级代码。正确分配EC编号对于理解代谢、设计新药、工程化微生物以生产燃料或塑料替代品,以及追踪生态系统如何处理化学物质都至关重要。然而,用实验确定酶功能既缓慢又昂贵。本研究提出了HIT-EC,一种新的人工智能模型,能够从蛋白质序列可信地预测EC编号,并解释它为何给出该预测。
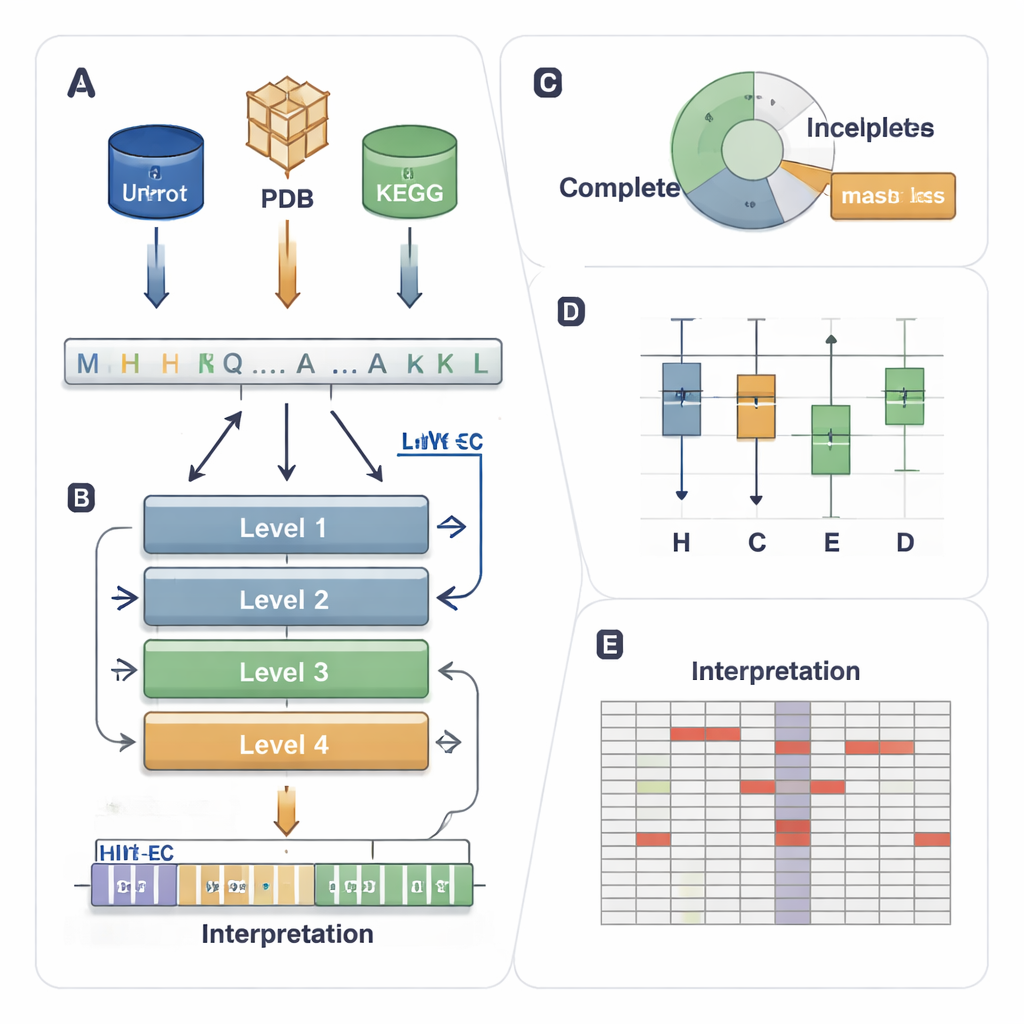
酶功能的邮编系统
EC系统为每个酶分配一个四级代码,例如1.1.1.37。第一个数字表示一个宽泛的类别(例如,移动电子或转移基团的酶),后面的数字描述更精细的反应细节。这种层次结构很有力,但也带来了苛刻的预测任务:模型必须在成千上万可能的代码中将四级都预测正确,即便有些酶很少见或在数据库中仅有部分注释(例如3.5.-.-,其中详细级别缺失)。现有计算方法使用3D结构、序列相似性或深度学习,但它们通常在处理罕见酶时表现欠佳,忽视部分标注的数据,并且通常是“黑箱”模型,很少解释为何做出某个判断。
遵循EC阶梯的四层人工智能
HIT-EC(用于EC预测的分层可解释变换器)构建上模仿了四步EC层级。它将原始蛋白序列输入,通过四个变换器层,每层专注于一个EC级别。局部通路将各级与上级连接,确保细粒度的决定(第四位)必须与更宽泛的决定(第一位和第二位)一致。并行地,全局通路在每一步保持完整序列上下文可见。模型还可以在具有不完全标签的序列上训练,使用“掩码损失”简单地忽略缺失的EC级别,而不是丢弃该序列。这使得HIT-EC能够从经人工整理的数据库中大量只做部分注释的蛋白质中学习。
在准确性和速度上胜过对手
作者汇集了一个大规模、经过仔细过滤的数据集,包含约200,000个酶,涉及1,938个不同的EC编号,来源于Swiss-Prot和蛋白质数据银行。在反复的留出测试中,HIT-EC在整体和按类F1评分上均超过了三种领先方法(CLEAN、ECPICK和DeepECtransformer),F1评分衡量了正确命中与误报之间的平衡。它在样本数少于或等于25个已知实例的低代表性EC编码上表现尤其强劲,这是先前方法常见的弱点。HIT-EC在训练后对新加入Swiss-Prot的酶以及来自不同细菌的完整基因组(包括研究透彻的大肠杆菌、枯草芽孢杆菌和结核分枝杆菌菌株)也具有良好泛化能力。尽管架构复杂,该模型效率很高:在标准GPU上处理单个蛋白质大约需要38毫秒——这比依赖较慢相似性搜索或多个模型集成的一些竞争方法快数十倍。
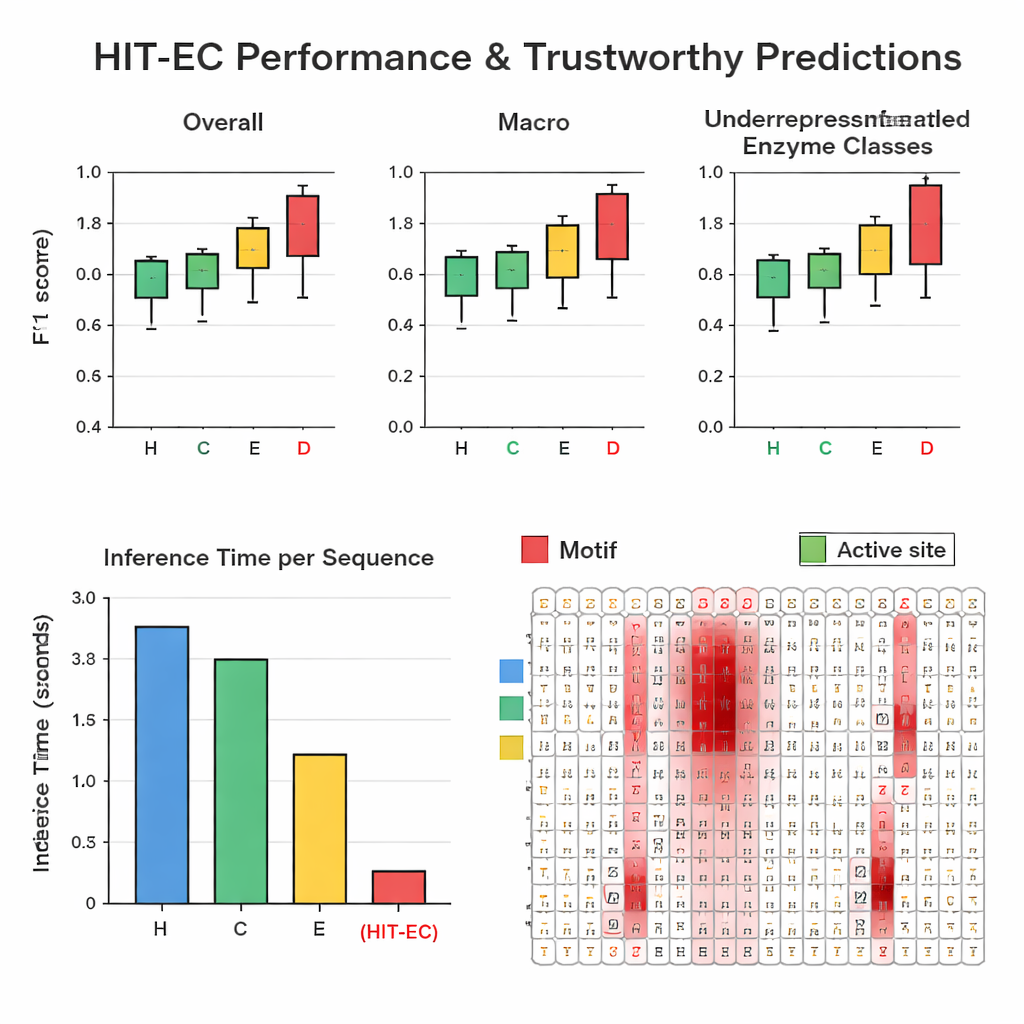
可视化模型“关注”的位置
为使预测可信,HIT-EC设计为展示序列中哪些氨基酸影响了每个EC级别的决策。作者构建了一条解释路径,将注意力权重与梯度信息结合起来,对每个位置的重要性进行打分。他们在表征良好的酶家族上验证了这些得分。例如,在一个细胞色素P450家族(CYP106A2)中,HIT-EC突出显示了已知的功能基序,如结合氧和结合血红素的区域,并识别出一个基准模型漏掉的细微EXXR基序。对于每个顶层EC类的经典代表——如酒精脱氢酶、己糖激酶和碳酸酐酶——模型的相关性得分都点亮了课本上记载的特征基序和底物结合位点。这些解释提供了生化层面的“证据”,表明模型的判断基于有意义的特征,而非偶然相关性。
指导对罕见与新兴酶的研究
团队进一步在两个对污染清理重要但研究不足的酶上测试了HIT-EC:一个参与降解芳香族污染物的细胞色素P450,以及来自链霉菌的降解PET的水解酶,后者有助于消化与塑料有关的分子。这两种酶都已被实验表征但缺乏官方EC分配。HIT-EC正确预测了预期的EC编号,并突出了与结构和生化研究已知信息相符的基序模式和催化残基。总体而言,这项工作表明HIT-EC不仅能比现有工具更准确、更快速地分配EC编号,尤其在罕见功能方面表现突出,还能解释为何认为某个酶执行特定化学任务。这种性能与可解释性的结合使其成为基因组学、生物技术和环境研究中进行大规模、可靠酶注释的有前景的工具。
引用: Dumontet, L., Han, SR., Lee, J.H. et al. Trustworthy prediction of enzyme commission numbers using a hierarchical interpretable transformer. Nat Commun 17, 1146 (2026). https://doi.org/10.1038/s41467-026-68727-3
关键词: 酶功能预测, 生物学中的深度学习, 变换器模型, 蛋白质注释, 生物修复酶