Clear Sky Science · zh
通过迁移学习改善代表性不足群体的多基因评分预测
为什么你的 DNA 风险评分可能不适用于你
基因“风险评分”越来越多地用于估计一个人患常见疾病(如糖尿病、心脏病或高血压)的可能性。但这些评分大多基于欧洲血统人群的 DNA 数据。因此,它们经常对来自其他背景的人预测不准确,引发关于公平性和临床实用性的担忧。本研究提出一个简单的问题:我们能否在不共享任何原始数据的情况下,重用从大型欧洲数据集中学到的知识,来为代表性不足的群体构建更好、更公平的基因评分?
从数百万个 DNA 标记到一个风险评分
多基因评分像是一张成绩单,将分布在全基因组上许多遗传标记的微小效应相加。每个标记都有一个权重,反映它与某一性状的关联强度,这些权重来源于大型遗传学研究。当这些研究以欧洲人为主时,得到的评分往往在欧洲人群中表现最佳。遗传背景的差异——某些 DNA 变体的频率以及它们的连锁继承模式不同——导致相同的权重在非裔美国人、西班牙裔和其他人群中往往失准。为每个群体收集同等规模的数据既昂贵又缓慢,因此作者采用了一种称为迁移学习的机器学习策略:不是在每个群体都从零开始,而是对在其他地方训练的现有模型进行改进。
如何在不共享原始数据的情况下借用知识
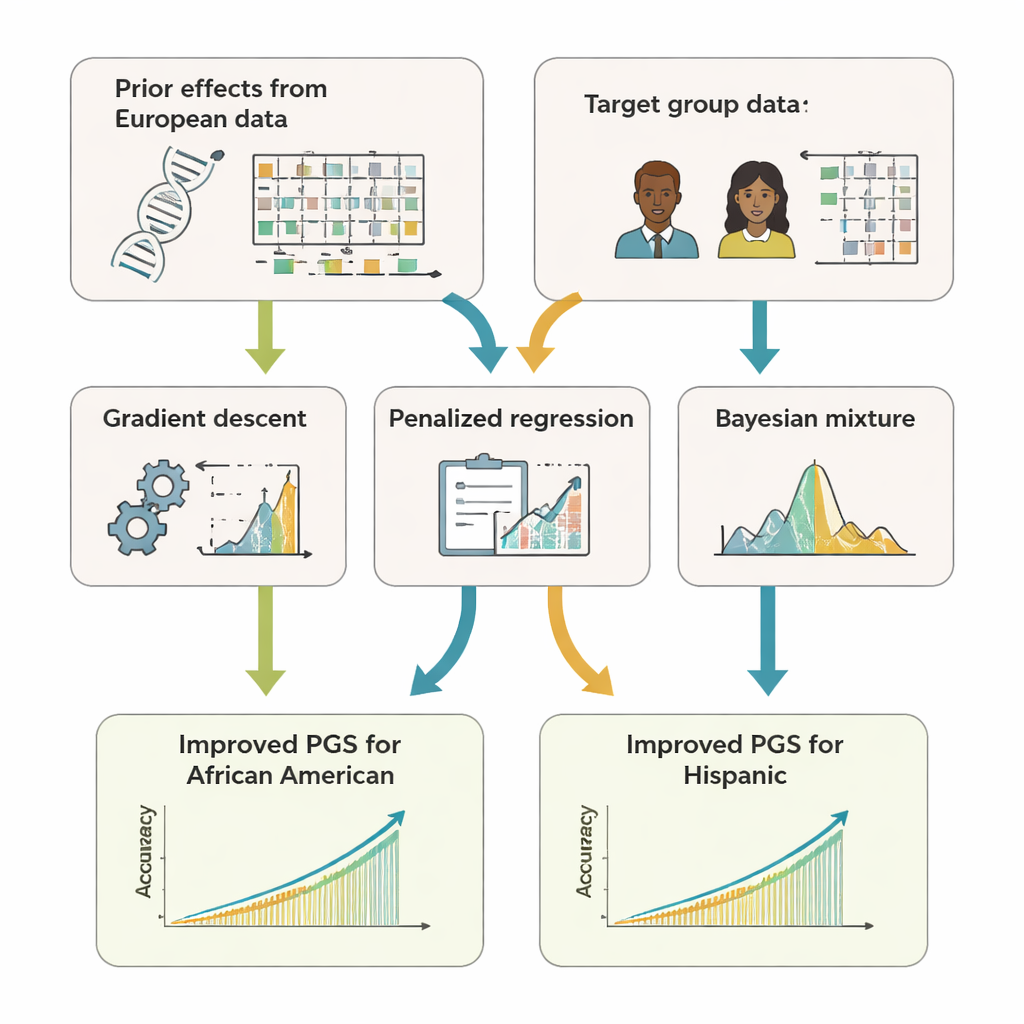
研究团队开发了 GPTL,这是一个开源的 R 软件包,实施了三种用于基因评分的迁移学习方法。三种方法都以在大型欧洲血统数据集中得到的 DNA 效应估计为起点,然后使用目标群体(例如非裔美国人或西班牙裔)数据对这些估计进行温和调整。第一种方法通过梯度下降逐步微调欧洲权重,并提前停止,以避免完全覆盖原始权重。第二种方法称为惩罚回归,它在没有足够证据时会将新估计主动拉向原始值。第三种方法是贝叶斯混合模型,允许每个 DNA 标记在多种信息源之间选择——例如来自不同血统群体的估计,甚至“无效应”选项——并根据这些信息对目标数据的解释优劣进行加权融合。
将方法付诸检验
为了评估这些方法的效果,作者使用了计算机模拟以及来自英国生物样本库和美国 All of Us 研究计划中数十万名志愿者的真实数据。他们以非裔美国人和西班牙裔参与者为目标群体,以欧洲血统数据作为主要先验信息来源。针对包括身高、体重指数、血脂、血压和肾脏指标在内的 11 个性状,迁移学习生成的评分通常优于仅在目标群体内部构建的评分或直接沿用欧洲评分的结果。它们的准确性常常与或略高于那些需要合并多个群体原始数据的更复杂“多血统”方法。关键在于,GPTL 的方法只需要汇总统计数据——关于基因效应的聚合数字——因此机构可以在不暴露个体级基因记录的情况下合作。
更多 DNA 并非总是更好
研究人员还考察了如何最好地选择要包含的遗传标记。与常见的“使用所有可用标记总是有利”的观念相反,他们发现对于非裔美国人,尤其是西班牙裔群体,包含数以百万计的极弱信号反而可能损害性能,尤其是在使用高度简化的遗传相关表示时。聚焦于证据更充分的标记并使用更丰富的关于变体共遗传(连锁)信息通常能产生更准确的评分。研究还表明,加入来自多个人群的先验信息并谨慎建模群体间差异,可进一步提高预测能力。
这对更公平的基因风险预测意味着什么
对于非欧洲人群,现成的基因风险评分可能表现远不如在欧洲人群中,从而可能加剧健康不平等。该研究表明,迁移学习——用代表性不足群体的适度数据对现有欧洲基础评分进行智能微调——可以缩小这种差距。在实践中,这意味着医疗系统和研究者可以在不跨机构或族群合并原始数据的情况下构建更准确、更公平的基因工具,从而缓解隐私顾虑。虽然没有一种方法适用于所有性状和群体,GPTL 工具包表明,只要我们把过去的模型视为可调整的起点而非固定产物,更公平的基因预测在技术上是可实现的。
引用: Wu, H., Pérez-Rodríguez, P., Boehnke, M. et al. Improving polygenic score prediction for underrepresented groups through transfer learning. Nat Commun 17, 1973 (2026). https://doi.org/10.1038/s41467-026-68696-7
关键词: 多基因风险评分, 迁移学习, 基因预测, 健康差距, 群体遗传学