Clear Sky Science · zh
多国基因组分析强调非洲霍乱的区域性传播
追踪跨境病原体的重要性
霍乱每年仍在非洲使成千上万的人生病并死亡,但关于疫情如何起始、如何在国家间传播以及为何反复发生的基本问题仍未完全解答。本研究汇集了来自七个非洲国家的科学家和公共卫生团队,通过读取致病细菌的基因序列来追踪霍乱弧菌。研究人员比较了数百个细菌基因组,展示了近期霍乱如何跨越国界流动、不同株系在何处流行,以及这些知识如何有助于更精准地遏制未来疫情。
从大陆尺度审视霍乱
为超越零散的病例报告,非洲疾病预防控制中心发起了名为非洲霍乱基因组联盟(CholGEN)的合作。喀麦隆、刚果民主共和国、马拉维、莫桑比克、尼日利亚、乌干达和赞比亚的实验室测序了763个高质量的霍乱弧菌O1基因组,样本主要来自2019年至2024年间。这是迄今在非洲本土产生的最大规模霍乱基因组集合。将这些新基因组与此前来自非洲和亚洲近1,800个已测序样本并列分析后,研究团队得以重建近期非洲疫情在长期全球霍乱大流行中的位置。
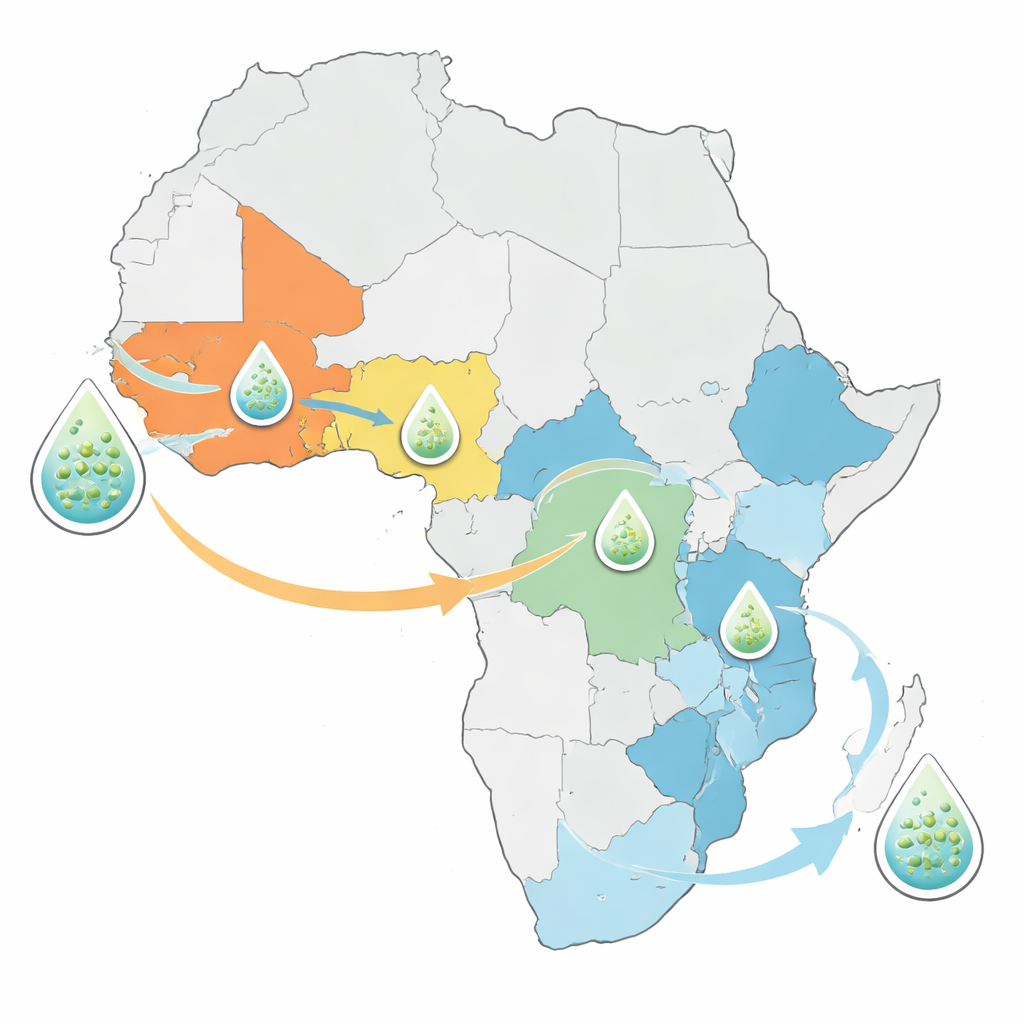
旧株系,新路线
分析显示,导致当前非洲疫情的霍乱株系并非全新的威胁。相反,所有新测序的细菌都来源于已知的第七次大流行霍乱谱系先前于1970年从亚洲传入非洲的引入事件。西非和中非国家如尼日利亚、喀麦隆和刚果民主共和国通常由一两个长期存在的谱系主导,这些谱系已持续数十年。相比之下,东非和南部非洲国家同时存在多种谱系的混合。其中一个被称为AFR15的谱系在近年迅速传播,与马拉维、赞比亚及邻国的异常大规模暴发有关,也与中东和南亚部分地区的流行有关。
疫情规模并非仅由基因决定
人们可能会怀疑AFR15的爆发性传播是由于其发生了重大遗传变异,使其更具危险性或更能逃避免疫与治疗。然而,当研究人员比较多条活跃谱系的突变速率和模式时,并未发现显著差异。细菌的进化速度相似,突变的类型和受影响的基因在各谱系间也大致相同。总体的抗生素耐药基因谱随时间和在各国之间也大体稳定。主要的例外是乌干达,该处细菌获得了一个携带多种耐药基因的大型可移动DNA元件(质粒),很可能随与也门和黎巴嫩相关的暴发株系一同引入。即便如此,研究并未发现能够单独解释近期非洲暴发严重性的新增基因。
更完善的取样揭示隐匿的传播轨迹
由于细菌基因组记载了近缘株系发现的位置,研究团队能够推断霍乱跨境的频率。他们检测到许多邻国间的国际传播实例,包括赞比亚与刚果民主共和国之间反复的交流。然而,进一步分析显示,跨境传播的统计信号在样本取样密集的年份和地区最为明显。这表明霍乱在现实中的移动可能比现有数据显示的更频繁;许多传播事件可能因为那些时间或地点没有进行基因测序而被忽视。为应对这一点,作者们建立了一个框架,用以估算一个国家通过测序更多样本能获得多少新信息,综合考虑遗传多样性、引入次数和已有数据量。
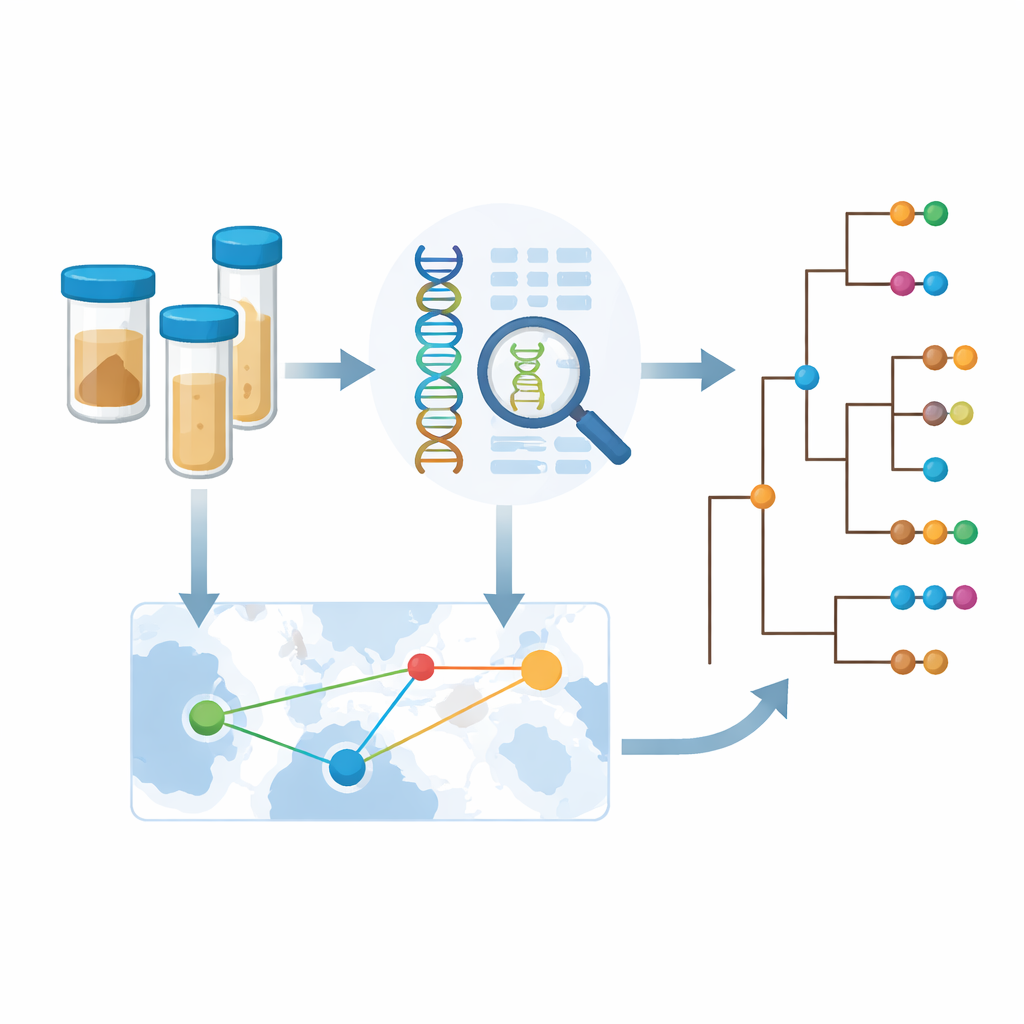
用基因组学指导更聪明的防控策略
研究得出的结论是,就当前非洲的霍乱而言,疾病如何以及在哪里传播比细菌本身的任何剧烈变化更为重要。研究结果支持例行且区域协调的基因组监测,使邻国能够及早发现共同的暴发、追溯来源,并更高效地针对干预措施(如疫苗接种活动、供水与环境卫生改善以及边境地区应对)进行部署。通过在非洲公共卫生实验室内部建立测序能力并跨境共享数据,像CholGEN这样的倡议为利用现代遗传学推动在2030年前将霍乱作为公共卫生威胁消除的宏伟目标提供了切实可行的蓝图。
引用: Mboowa, G., Matteson, N.L., Tanui, C.K. et al. Multicountry genomic analysis underscores regional cholera spread in Africa. Nat Commun 17, 2539 (2026). https://doi.org/10.1038/s41467-026-68642-7
关键词: 霍乱, 基因组监测, 非洲, 跨境传播, 抗菌素耐药性