Clear Sky Science · zh
在显式考虑未折叠态下评估蛋白质折叠稳定性
为什么蛋白质稳定性重要
你体内的每种蛋白质都是微小的分子机器,必须折叠成精确的三维形状才能正常工作。如果这种折叠太脆弱,蛋白质可能发生功能失常、聚集,甚至根本无法被生成——这些问题与疾病以及蛋白质类药物和酶研发中的失败有关。在实验室中测定蛋白质的稳定性既慢又复杂,因此科学家们正寻求计算方法,能够仅凭序列可靠地预测蛋白质的解折叠难易程度。
对折叠态与未折叠态的新审视
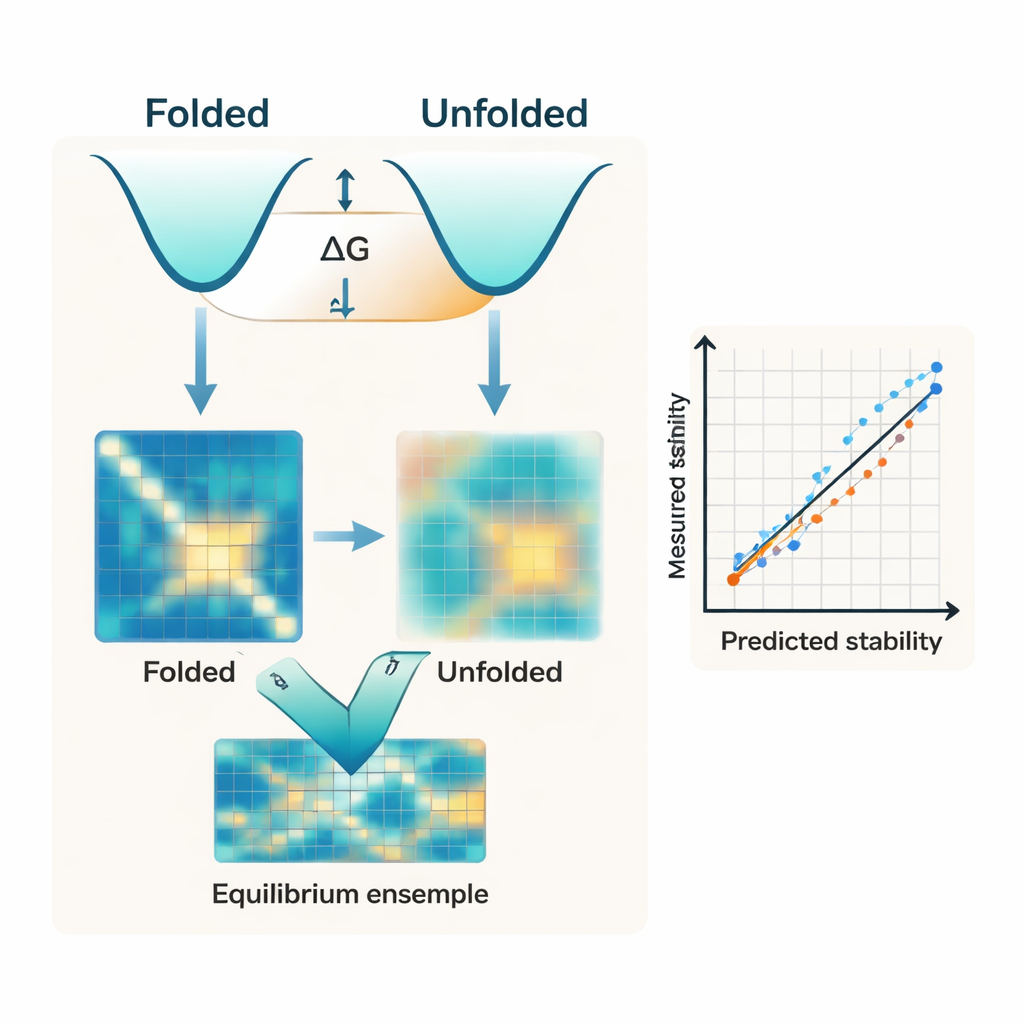
大多数现代算法几乎完全侧重于蛋白质的折叠形态。它们通常以诸如 AlphaFold 之类的 AI 预测结构为起点,将单一结构视为稳定性的主要决定因素。但稳定性实质上是两类广义集合之间的能量差:紧凑的折叠态和构成未折叠态的许多松散构象。作者指出,忽视未折叠一侧的平衡,是现有工具难以匹配实验折叠自由能(称为 ΔG)测量结果的关键原因之一。
一个同时学习两种态的新模型
研究者提出了 IFUM,一种用于估计 ΔG 的深度学习系统,同时学习每个蛋白质中折叠与未折叠态的平衡。IFUM 不把未折叠态当作模糊的背景,而是借鉴聚合物物理学的思想,将其表示为“随机线圈”,并将折叠态与未折叠态都编码为氨基酸对之间距离的图谱。该模型利用预训练的强大序列和结构网络的信息,联合预测整体稳定性以及描述在每对残基上有多少蛋白质群体处于折叠与未折叠状态的概率图。用一个包含大量小型、实验表征蛋白质和已知无序蛋白的数据集进行训练,使 IFUM 能识别既有良好结构的序列,也有松散无序的序列。
更好的数值与更广的突变覆盖
在一组经过精心控制的小蛋白质数据集上测试时,IFUM 在预测实验 ΔG 值方面比那些仅依赖折叠结构或只在序列上训练的语言模型具有更低的误差和更高的相关性。关键在于,该模型还能处理多种序列变体。它准确捕捉单点和双点突变的影响,以及改变蛋白质长度的插入与缺失——这些情形是许多现有工具要么直接失效,要么根本未被设计来处理的。内部比较显示,移除未折叠态目标会显著降低性能,这强调了显式建模未折叠集合不仅仅是一个概念上的改进,而是实现准确预测的核心要素。
从设计台到真实世界测试
为检验 IFUM 能否指导实际的蛋白质工程,作者将其应用于三个具有挑战性的设计问题:稳定干扰素‑λ(interferon‑lambda)蛋白、重塑免疫信号蛋白 IL‑10,以及改良一种名为 UGT76G1 的糖类修饰酶。在这三种案例中,IFUM 预测的稳定性与测得的熔解温度(反映蛋白质在加热下解折叠的耐受程度)吻合良好。该模型还帮助筛选数百个全新计算设计的蛋白,挑选出最有可能折叠并在细胞中保持可溶的候选,表现优于结构预测网络中常用的置信度评分。这些结果表明,IFUM 可作为现代蛋白质设计工作流程中与基于结构的检查并行使用的实用“稳定性过滤器”。
局限与未来方向
像任何模型一样,IFUM 也有边界。它主要在短链、单链、可溶蛋白上训练,对于更大尺度的蛋白、具有广泛柔性环区或跨膜区的蛋白,其绝对稳定性数值变得不那么可靠。其对未折叠态的描述仍是简化的统计模型,而非对所有可能构象的完全逼真刻画。尽管如此,这种方法展示了让 AI 同时考虑折叠与未折叠集合能够产生更可靠的稳定性估计。对于非专业读者,关键结论是:IFUM 使我们更接近能用定量置信度问计算机“这个蛋白设计真的能保持结构吗?”,从而有望加速更安全的生物药物和更稳健的工业酶的开发。
引用: Lee, H., Cho, Y., Yun, J. et al. Protein folding stability estimation with explicit consideration of unfolded states. Nat Commun 17, 1883 (2026). https://doi.org/10.1038/s41467-026-68637-4
关键词: 蛋白质稳定性, 蛋白质折叠, 深度学习, 蛋白质设计, 突变