Clear Sky Science · zh
对EGFR抑制剂产生耐药性的肺癌对一种共价、非半胱氨酸依赖的使KEAP1聚合的分子桥表现出伴随敏感性
为何耐药性肺癌重要
靶向药物通过针对一种称为EGFR的异常生长信号,已经彻底改变了某些肺癌的治疗方法。然而对大多数患者来说,这类药物在几年内会因癌症进化出耐药性而失效。本研究揭示了一个令人意外的转折:一旦肿瘤对EGFR抑制剂产生耐药性,它们便会出现一种新的阿喀琉斯之踵,可通过另一类化合物加以攻击。理解这一隐性弱点可能会激发未来的治疗策略,转而围堵癌症进化,而不是始终被动追赶。
揭示的隐性弱点
研究者将注意力集中在由EGFR突变驱动的非小细胞肺癌,这是该病的一种常见形式。在实验室中,他们比较了对药物敏感的癌细胞与那些进化出对吉非替尼和奥西替尼等EGFR抑制剂耐药的密切相关细胞。随后他们筛测了约2100种小分子,观察哪些分子能比原始、对药物敏感的细胞更有效地杀死耐药细胞。在众多候选者中,一种名为MCB-613的化合物持续显现出显著效果。对EGFR抑制剂无动于衷的耐药细胞在细胞培养和小鼠肿瘤中都对MCB-613表现出异常脆弱。
困住混合肿瘤群体 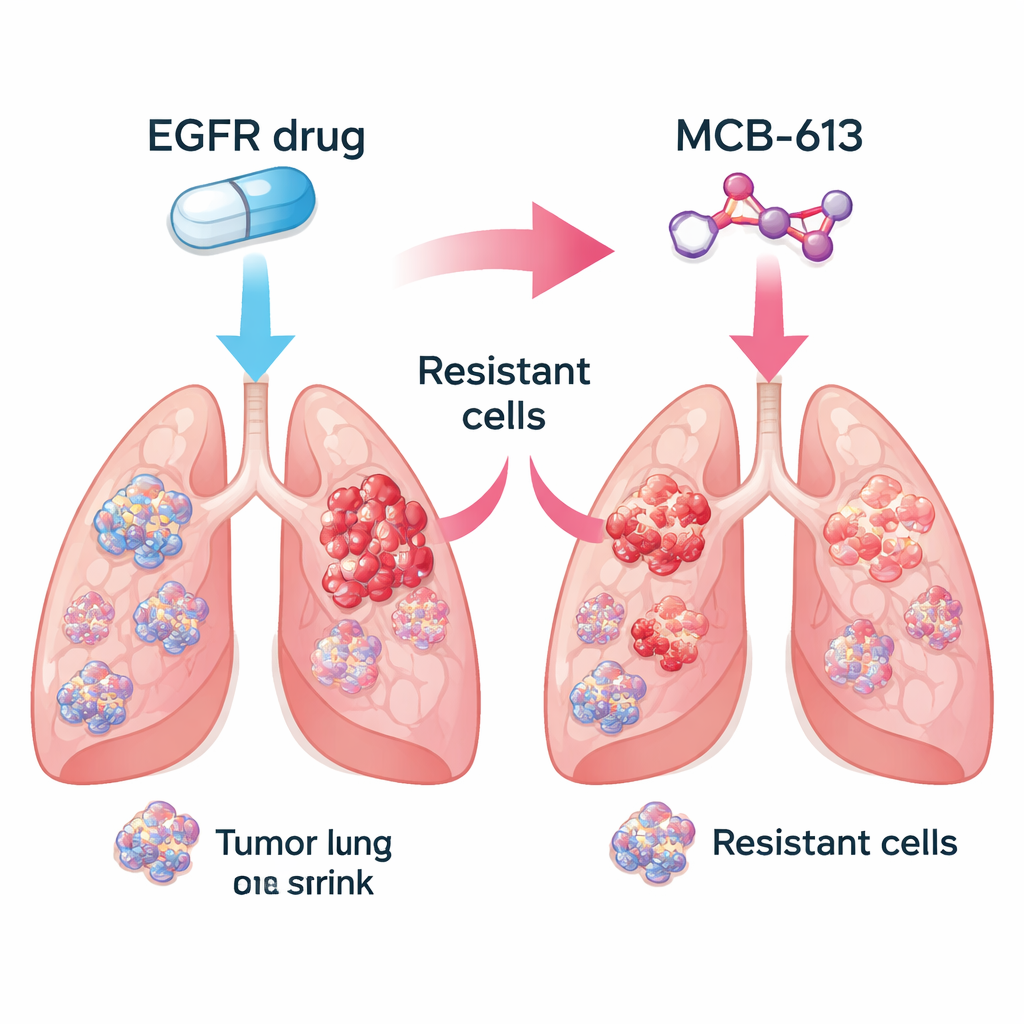
Figure 1.
真实的肿瘤由多种细胞混合而成:有些仍对原始药物敏感,而另一些通过不同的遗传策略获得耐药性。研究团队探究将EGFR抑制剂与MCB-613联合使用是否能消除这种多样性。在一项受控实验中,他们将大部分对药物敏感的细胞与一小部分多种耐药类型的细胞混合,模拟患者肿瘤。单独用EGFR抑制剂或MCB-613处理时,仍有细胞存活并生长。但两者联合使用时,整个细胞群体崩溃。这表明将标准靶向疗法与经过精心挑选的“伴随敏感性”药物配对,可能把肿瘤逼入进化的死胡同。
打破守护者的分子桥
为了解为什么MCB-613对耐药细胞打击如此严重,科学家检查了它结合的蛋白。通过化学探针和靶向CRISPR基因敲除筛选,他们将一种名为KEAP1的蛋白确定为MCB-613作用所必需。KEAP1通常充当细胞的守护者,感知应激并帮助调节保护性反应。团队发现MCB-613以一种不寻常的方式与KEAP1结合:它表现得像一座刚性分子桥,将KEAP1单元连接成过大且异常的聚集体。该过程并不依赖于KEAP1中常见的反应性含硫位点,而是依赖于其二聚化区域中的一个特定赖氨酸残基。当该赖氨酸被突变时,MCB-613无法再使KEAP1聚集,耐药细胞也不再对该化合物表现出超敏感性。
将有益应激变为致命过载 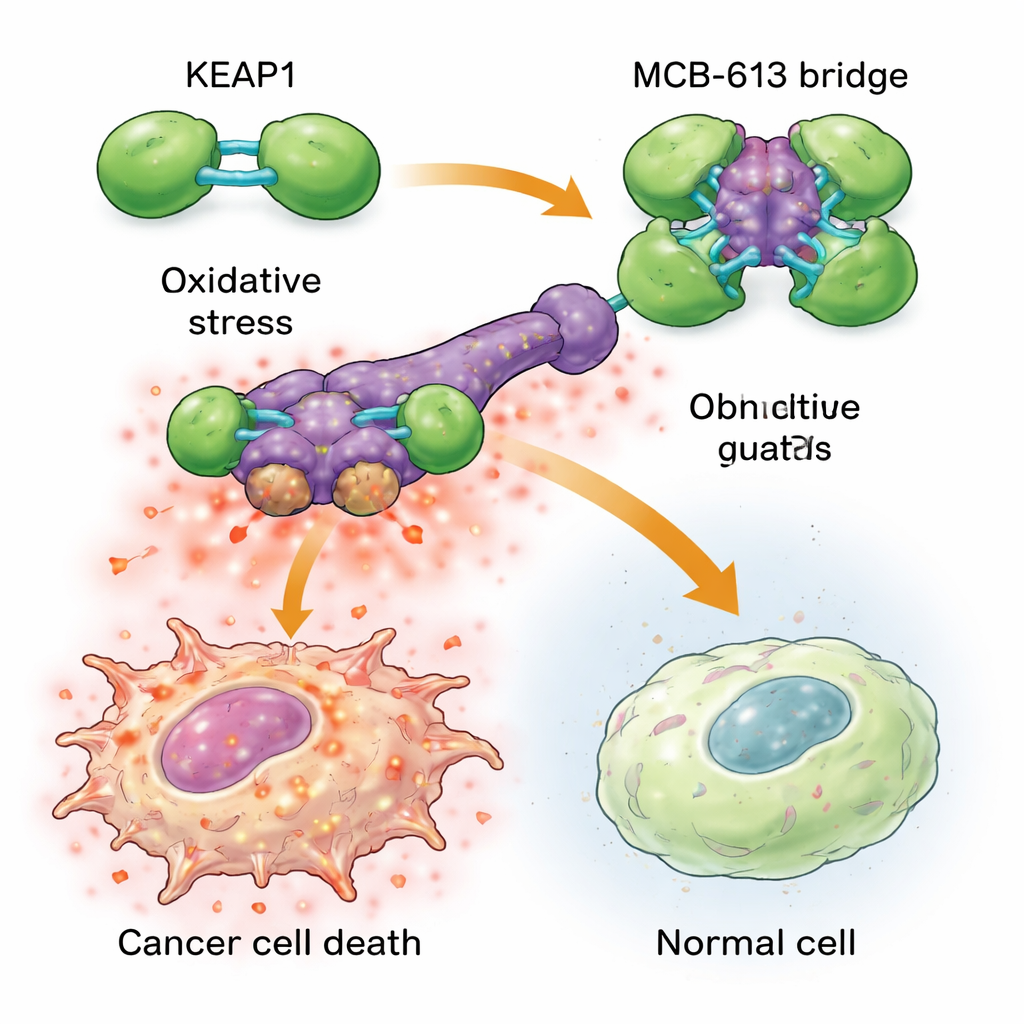
Figure 2.
KEAP1的聚集在耐药癌细胞内触发了一系列危险反应。这些细胞本已处于较高的背景应激水平,具有升高的活性氧(有害的化学副产物)水平以及整合应激反应这一保护性信号网络的增强活性。当加入MCB-613时,KEAP1受损将这种应激状态推向临界点:活性氧进一步积累,关键的应激调节因子ATF4和CHOP被激活,触发强烈的细胞死亡程序。阻断这些应激调节因子或用化学方法清除活性氧,在很大程度上可以保护细胞免受MCB-613的杀伤。有趣的是,经典的KEAP1伙伴NRF2——常被认为是抗氧化防御的主要驱动者——并不是导致细胞死亡的原因;事实上,去除NRF2反而使细胞更为敏感,这强调MCB-613利用的是一条不同的、非常规通路。
这对未来治疗的可能意义
目前,MCB-613本身只是一个具有化学缺陷的工具化合物,不适合作为药物。但它揭示了一个强有力的概念:随着肺癌对EGFR抑制剂进化出耐药性,它们可能陷入一种可被选择性靶向的应激状态——通过将KEAP1迫使形成功能失调的组装体来实现。原则上,此类“分子桥”的改良版本可以被开发得更安全、更精准,为肿瘤学家提供一种手段,使肿瘤在对原始靶向疗法敏感与对后续诱导应激剂敏感之间做出“无法选择”的抉择。这种进化困陷策略最终可能有助于延缓或克服EGFR突变肺癌以及其他难治癌症的耐药问题。
引用: Bassil, C.F., Dillon, K., Anderson, G.R. et al. EGFR inhibitor-resistant lung cancers exhibit collateral sensitivity to a covalent, cysteine-independent KEAP1 oligomerizing molecular bridge. Nat Commun 17, 1726 (2026). https://doi.org/10.1038/s41467-026-68424-1
关键词: EGFR突变的肺癌, 药物耐药, 伴随敏感性, KEAP1, 氧化应激