Clear Sky Science · zh
通过调节p53的乙酰化/磷酸化状态,ATGL使肝细胞癌细胞对致基因药物更敏感
将脂肪分解变为癌症弱点
肝癌的标准化疗常常失败,原因在于肿瘤细胞在应对DNA损伤方面异常擅长生存。本研究探讨了这些癌细胞内一个出人意料的盟友:一种分解储存脂肪的酶。通过增强这种称为ATGL的酶,研究者发现可以促使肝肿瘤细胞停止修复DNA损伤并走向自毁。该工作揭示了癌细胞处理脂质的方式与它们对强效致基因药物反应之间的隐藏联系,提示了使现有治疗更有效的新途径。
为何肝脏肿瘤能抵抗强效药物
肝癌,尤其是肝细胞癌,是全球最常见且致死率高的肿瘤类型之一。许多患者接受如依托泊苷和多柔比星等损伤DNA的药物,期望将癌细胞推入致命危机。然而这些细胞常通过停滞生长并激活由守护蛋白p53控制的修复系统而逃脱。如果损伤可以修复,细胞便恢复分裂;若不能,p53也能触发程序性细胞死亡。核心难题是哪些因素将p53的作用倾向于修复还是自毁,以及为何某些肿瘤对治疗仍然顽固地耐受。
一种脂肪切割酶改变了天平
研究团队将注意力集中在ATGL上,这是一种在称为脂滴的小细胞储罐中修剪储存脂肪的酶。在肝癌中,ATGL的水平通常低于健康组织,早期研究也提示强制表达更多ATGL会减缓肿瘤生长。本研究中,研究者对肝癌细胞系进行工程改造,使其过度表达或下调ATGL,然后用致DNA药物处理。过表达ATGL的细胞积累了更多的DNA断裂征象,而降低ATGL的细胞则显示出较少的损伤。用特异性抑制剂阻断ATGL的切割活性,或表达无法起作用的突变体,均消除了这种敏感性增强,证明该酶的脂肪分解活性本身是关键。
重接细胞的抉择:修复还是死亡
深入研究时,科学家们检查了p53,它在DNA损伤后起到分子交通警察的作用。p53的行为受添加到特定位点的小化学标记控制。在ATGL高表达的细胞中,致基因药物导致p53获得更多的一类标记(乙酰基),而相对较少另一类(磷酸基)。这一转变更有利于激活促进细胞死亡的基因,如Puma,同时抑制通常用于暂停细胞周期并支持DNA修复的基因,如p21和GADD45。结果是,即便药物被洗掉,ATGL高表达的细胞也未能清除DNA损伤标志,而是走向凋亡而非恢复。
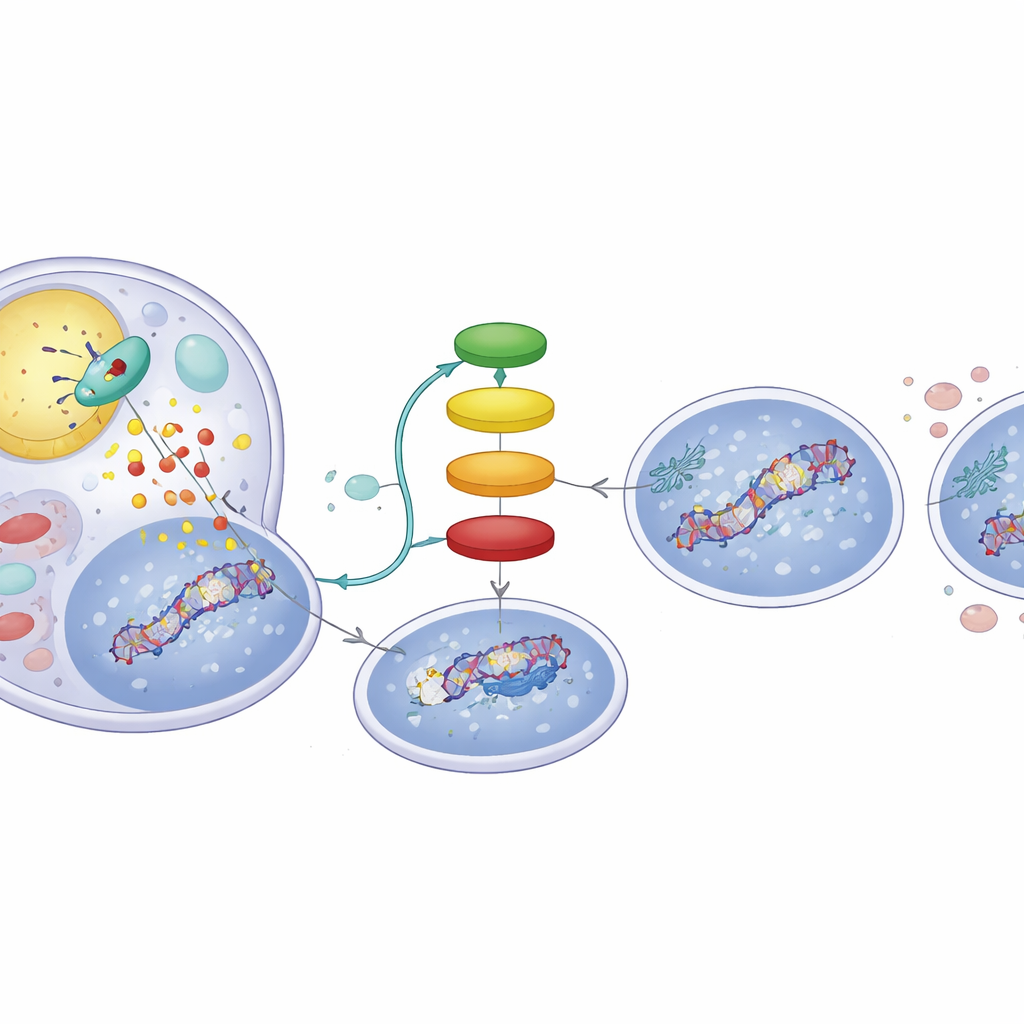
肿瘤细胞内的脂肪驱动信号链
脂肪的分解如何改变p53的标记?ATGL作用的分解产物是游离脂肪酸,它们可作为信使。研究表明这些脂肪酸激活了一个核受体PPARα,后者又增强了p300的活性——p300是一种将乙酰基添加到p53上的蛋白。当研究者使用PPARα激活化合物时,复制出了ATGL高表达时的行为:DNA损伤信号增加且p53朝向促凋亡的谱系。相反,阻断p300则抹消了ATGL诱导的p53变化并减少了DNA损伤,强调ATGL → PPARα → p300这条链对该开关至关重要。对数百例公开数据集中人类肝肿瘤的分析也呼应了这一联系,显示ATGL表达较高的肿瘤通常伴随更强的PPARα和p300特征以及p53控制基因的表达。
这对未来治疗意味着什么
简而言之,研究表明,当通过ATGL鼓励肝癌细胞燃烧储存脂肪时,它们更不倾向于修复化疗引起的DNA损伤,而更可能进入有序的细胞死亡。这提示了两个实际可能性:检测ATGL水平或可帮助预测哪些患者的肿瘤更易对致基因药物产生响应;同时,增强ATGL活性或其下游的PPARα通路可能与现有化疗联合使用以克服耐药。尽管仍需在动物和患者中进一步验证,该工作凸显了一个耐人寻味的信息:在肝癌中,使肿瘤细胞在微观层面上“更精瘦”或许也能让它们对挽救生命的药物更脆弱。
引用: Castelli, S., De Cristofaro, A., Desideri, E. et al. ATGL sensitizes hepatocellular carcinoma cells to genotoxic drugs by modulating p53 acetylation/phosphorylation status. Cell Death Discov. 12, 164 (2026). https://doi.org/10.1038/s41420-026-03048-4
关键词: 肝细胞癌, ATGL, DNA损伤反应, p53信号, 癌症中的脂质代谢