Clear Sky Science · zh
新型双胍类化合物4C对EGFR的抑制通过下调BRCA2和Rad51增强卵巢癌对PARP抑制剂的敏感性
这项研究为何重要
对于许多被诊断为卵巢癌的人来说,现有的靶向药物仅对携带特定遗传缺陷的少数肿瘤有效。本研究探索了一种将一类重要药物(称为PARP抑制剂)的益处扩展到更大患者群体的方法,即那些肿瘤缺乏这些突变的患者。研究者通过将一种新型实验性化合物与现有药物配对,展示了一种策略,可以将癌细胞推入致命的死胡同,同时尽可能保护健康组织。
当前卵巢癌治疗的一个障碍
卵巢癌常常在晚期才被发现,仍然是影响女性的最致命癌症之一。PARP抑制剂(如奥拉帕利)在某些病例中效果显著,但主要限于那些在DNA修复通路(与BRCA1和BRCA2基因相关)存在缺陷的患者。然而,大多数肿瘤仍保有完整的修复机制,能够修复这些药物造成的DNA损伤,从而让癌细胞存活。该修复体系的关键成员包括两种蛋白BRCA2和Rad51,它们通过同源重组过程修复断裂的DNA链。找到有选择性削弱癌细胞中该修复通路的方法,可能使PARP抑制剂对更多患者有效。
一个众所周知的生长开关的作用
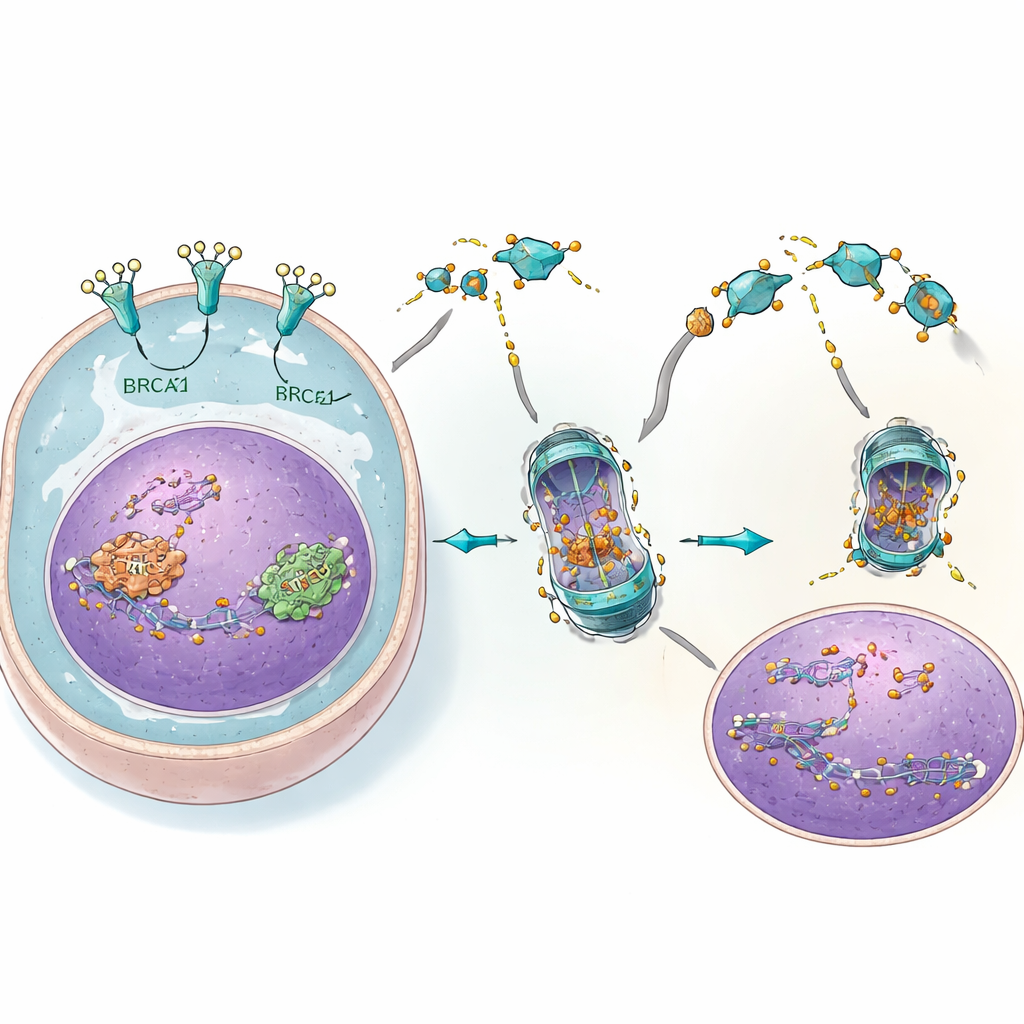
研究团队聚焦于一种熟知的癌症相关分子——表皮生长因子受体(EGFR),它位于许多肿瘤细胞表面并驱动其生长。他们发现EGFR不仅仅发送生长信号:在BRCA基因正常的卵巢癌细胞中,高水平的EGFR与患者生存率较低和对PARP抑制剂的耐受相关。当在细胞培养和小鼠体内降低或关闭EGFR时,肿瘤对奥拉帕利的敏感性显著提高。研究者显示,EGFR在DNA损伤后协助将BRCA2和Rad51送入细胞核,在那里它们修复断裂并削弱治疗效果。仅仅抑制EGFR的酶活性并不足以达到这一目的;必须降低EGFR蛋白的总体含量,才能显著扰乱该修复通路。
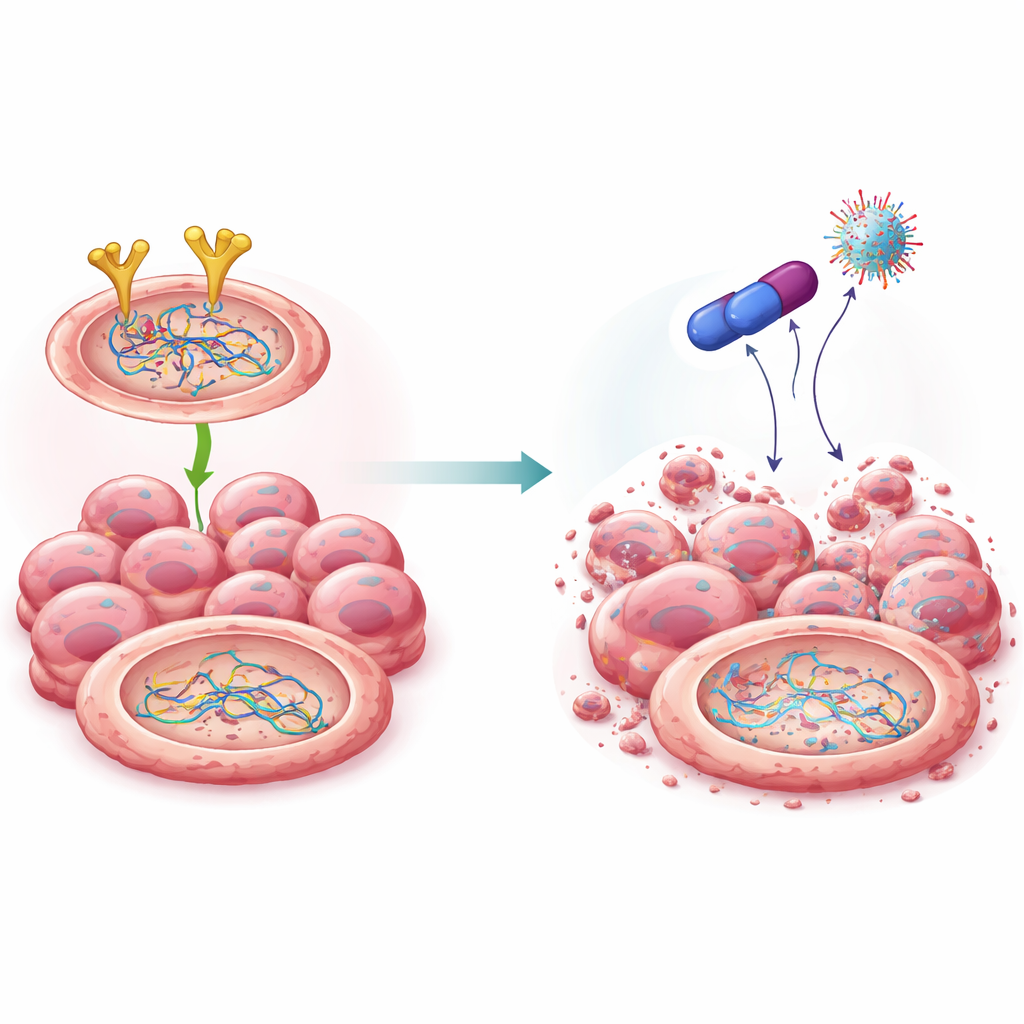
一种使修复团队失能的新化合物
在早期关于双胍类药物家族的工作基础上,科学家合成了若干同系化合物,并鉴定出一种突出分子,称为4C。该化合物对卵巢癌细胞呈强烈毒性,而对正常细胞相对温和。计算机建模和体外实验表明,4C可直接与EGFR结合并将其标记送入细胞的蛋白质降解机制。与一些现有的EGFR药物不同,4C降低的是EGFR的整体量而不仅仅是抑制其活性。随着EGFR水平下降,BRCA2和Rad51的稳定性也随之降低:它们被标记、分解,无法再支持高效的DNA修复。重要的是,这种削减发生在蛋白水平,而并未改变其对应的基因。
阻断细胞内的救援通路
研究揭示了连接DNA损伤与肿瘤存活的更详细事件链。在PARP抑制剂造成DNA损伤后,另一种感应蛋白ATM会从细胞核向胞体发送信号。作为响应,EGFR与BRCA2和Rad51联手,帮助它们进入细胞核以修复损伤。研究者发现第三种蛋白c-Cbl通常作为BRCA2和Rad51的“处置标签酶”。EGFR与c-Cbl竞争接触这些修复蛋白,从而保护它们不被标记为降解目标。当4C降低EGFR水平时,c-Cbl更易与BRCA2和Rad51结合,导致它们被标记、降解并丧失功能。核内可用的修复蛋白减少后,DNA损伤累积,癌细胞对PARP抑制剂的敏感性显著提高。
从细胞到动物:一种强效组合
在细胞培养和小鼠模型中,将4C与PARP抑制剂联合使用产生了强力的协同效应。对单独使用任何一种药物都有抗性的、BRCA基因正常的肿瘤在两者联合处理下显著缩小或停止生长。DNA损伤的标志大幅上升,支持修复被压垮的观点。与此同时,正常细胞及肝、肾等关键器官几乎未见明显损害,这可能是因为它们的EGFR水平较低且较少依赖该特定修复捷径。该组合的益处还包括减少卵巢癌细胞在体内的扩散。
这对患者可能意味着什么
这项工作提出了一种将癌细胞优势转化为弱点的方法。通过用新型化合物4C靶向EGFR,研究者剥夺了BRCA2和Rad51的保护,使那些本来DNA修复正常的肿瘤在应对PARP抑制剂时表现得更像BRCA突变肿瘤。这种被称为“合成致死”的强制性脆弱性,可能将现有药物的适用范围扩大到更多卵巢癌患者,同时保持副作用在可控范围内。尽管4C仍处于实验阶段,需要在更高级别模型和临床试验中进行大量测试,但该研究为如何通过拆解肿瘤细胞中特定修复帮手来改善治疗效果提供了清晰的蓝图。
引用: Xiao, D., Yao, J., Yang, X. et al. Repression of EGFR by new biguanide 4C potentiated ovarian cancer to PARP inhibitors through down-regulation of BRCA2 and Rad51. Cell Death Dis 17, 317 (2026). https://doi.org/10.1038/s41419-026-08556-w
关键词: 卵巢癌, PARP抑制剂, EGFR, DNA修复, 靶向治疗