Clear Sky Science · zh
PGC-1α通过Tim23依赖性抑制DRP1介导的铁死亡,从而保护免受MASH损害
这对日常健康为何重要
许多患有肥胖或2型糖尿病的人会在不知不觉中出现一种严重的肝脏问题,称为代谢功能紊乱相关脂肪性肝炎(MASH)。在这种情况下,充满脂质的肝细胞发生炎症并开始死亡,进而导致瘢痕形成、肝硬化乃至肝癌。该研究揭示了肝细胞内一个隐藏的自我保护系统——以线粒体为中心——它可以保护肝脏免受损伤,或在系统失效时加速疾病进展。理解这一内部安全开关,可能为全球最常见的肝脏威胁之一开辟新的治疗途径。
近距离审视这种隐匿的肝病
MASH发生于简单性脂肪肝转向更危险的状态时,表现为肝细胞肿胀受损、炎症及最终瘢痕组织。作者检查了来自MASH患者的肝组织样本和喂以高脂、高糖或营养缺乏饮食以模拟人类病情的小鼠模型。研究聚焦于一种称为铁死亡的特定细胞死亡方式,在此过程中铁与受损脂质相互作用,生成对细胞膜造成穿孔的有毒分子。在人类和小鼠的MASH肝脏中,肝细胞显示出这种由铁和脂质驱动的细胞死亡特征:过量铁沉积、畸形的线粒体以及促进脂质损伤的蛋白水平升高,同时正常清除有害副产物的蛋白水平降低。
证据表明阻断铁驱动的细胞死亡有益
为检验铁死亡是旁观者还是疾病驱动者,研究人员对高脂饮食小鼠给予专门阻断铁死亡的化合物ferrostatin-1。接受阻断剂的小鼠肝脏脂肪堆积减少、铁过载减轻,炎症和瘢痕的迹象也更少。血液检测显示肝功能改善和代谢健康好转,包括胆固醇下降和胰岛素敏感性提高。在用硬脂酸(一种模拟MASH中过量脂肪的脂肪酸)处理的离体小鼠肝细胞中,相同的药物减少了脂质积聚、铁负荷、氧化损伤和炎症信号。综合这些结果表明,铁死亡是MASH损伤的关键发动机,打断该过程可以显著缓解疾病。
线粒体内肝脏的内建守护者
研究小组进一步聚焦于PGC-1α,这是一种帮助线粒体产生能量并应对压力的主调控因子。在人类MASH肝脏、病变小鼠和受压的肝细胞中,PGC-1α水平明显降低,而一种线粒体分裂蛋白DRP1和一种激活脂质的酶ACSL4水平升高。使用仅在肝细胞中缺失PGC-1α的基因工程小鼠,作者发现失去这一守护因子使高脂饮食造成的损害大为加重:肝脏脂肪更多、炎症更重、铁负荷更高,并表现出更强的铁死亡信号。在细胞层面,PGC-1α缺失增强了DRP1活性,增加了ACSL4和铁输入蛋白,并削弱了通常抑制铁死亡的抗氧化防御。
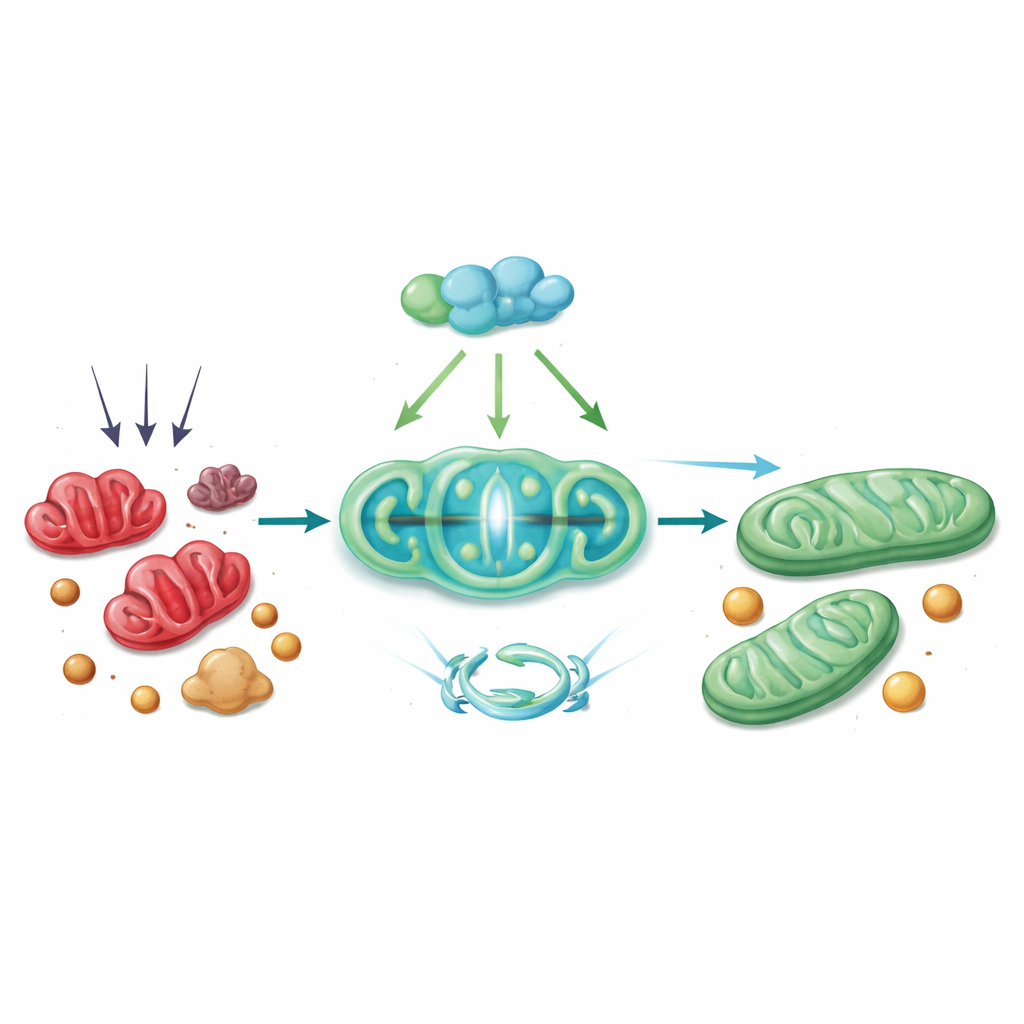
细胞内的一条保护性连锁反应
在机制上,PGC-1α通过一系列伙伴发挥作用。它与转录因子Nrf1协同,提升Tim23的产生——Tim23是内线粒体膜上的一个通道,对蛋白质导入和维持健康结构至关重要。当Tim23水平下降时,线粒体膜电位受损,会触发DRP1使细胞器片段化。研究显示,Tim23减少时,DRP1更加活跃并更容易在线粒体表面与ACSL4结合,将这一脂质修饰酶招入线粒体。在那里,ACSL4助长了那些使细胞易于发生铁死亡的脂质改变。通过在小鼠中使用病毒载体恢复PGC-1α或在培养的肝细胞中用基于CRISPR的激活器恢复PGC-1α,逆转了这些步骤:Tim23上调,DRP1和ACSL4活性下降,线粒体更为健康,铁死亡和肝损伤的标志物减少。
这一发现如何指导未来疗法
对非专业读者而言,主要结论是肝脏具有针对铁和脂质驱动细胞死亡的内部刹车装置,而这套刹车装置内置于线粒体中。PGC-1α–Tim23–DRP1–ACSL4这条链路像一套安全电路:当PGC-1α充足时,Tim23维持线粒体稳定,DRP1和ACSL4受到抑制,肝细胞不太可能自毁;当这套电路失效时,铁死亡加速,MASH恶化。通过在人类组织和动物模型中识别出这一通路,研究提出了两条互补的未来治疗策略——直接阻断铁死亡,或增强PGC-1α或Tim23活性以稳定线粒体——为在不可逆肝脏瘢痕形成之前实现更早且更有效的干预带来希望。
引用: Zhao, Y., Zhang, L., Li, B. et al. PGC-1α protects against MASH via Tim23-dependent inhibition of DRP1-mediated ferroptosis. Cell Death Dis 17, 246 (2026). https://doi.org/10.1038/s41419-026-08493-8
关键词: 脂肪肝疾病, 线粒体, 细胞死亡, 铁代谢, 肝脏炎症