Clear Sky Science · zh
环鸟苷酸-蛋白激酶G信号通过ULK1介导的自噬减轻主动脉瓣钙化
为什么心脏瓣膜“生锈”很重要
随着年龄增长,主动脉瓣——心脏的主要出流闸门——会逐渐变硬并沉积钙质,这种情况称为钙化性主动脉瓣疾病。瓣膜“硬化”迫使心脏更费力地泵血,可能导致胸痛、晕厥、心力衰竭,或需要置换瓣膜手术。目前尚无可靠减缓这一过程的药物;临床上只能等到瓣膜严重受损再行手术。本文研究了一条内在细胞通路,该通路似乎能保护瓣膜免于钙化,并检验了一种现代心衰药物作为可能重新激活这一路径的手段。
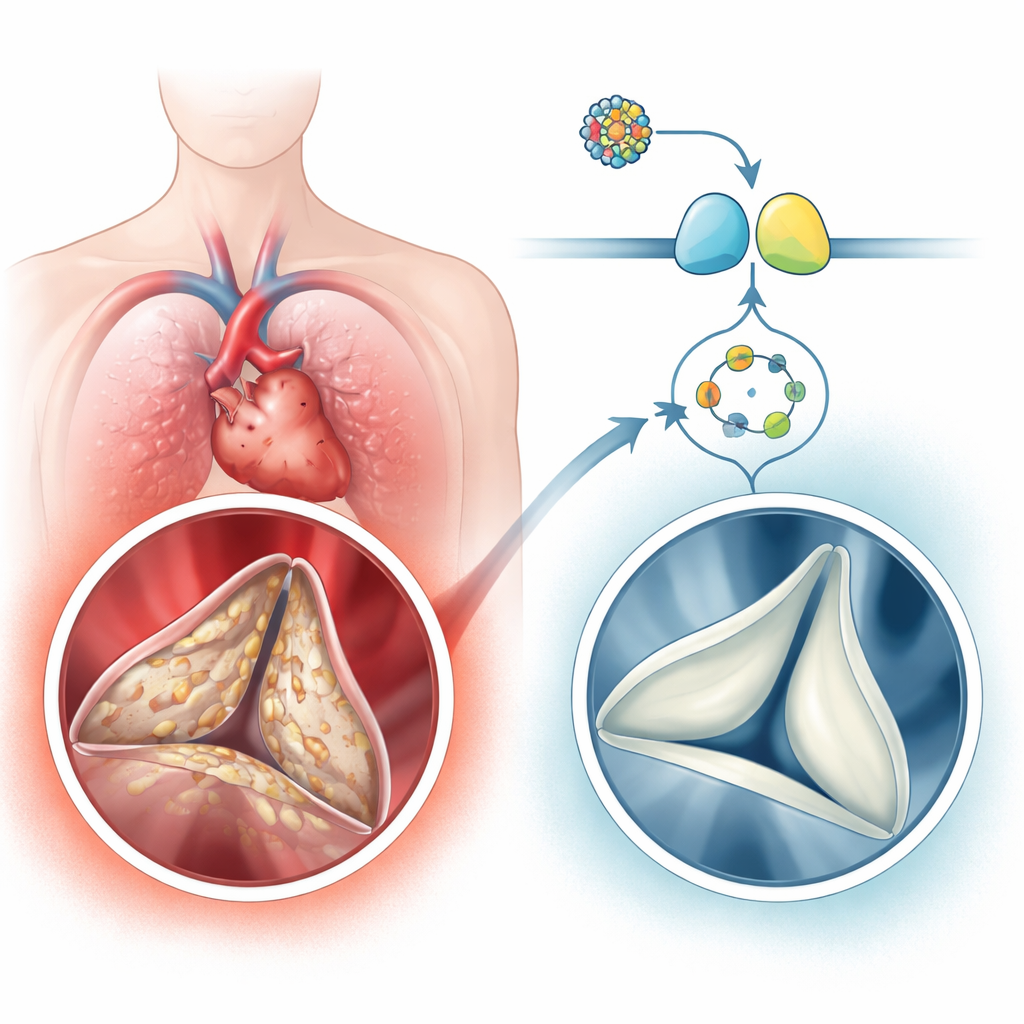
一种常见但常被忽视的心脏问题
钙化性主动脉瓣疾病在老年人中出人意料地常见,影响65岁以上人群的数个百分点,在75岁以上人群中高达十分之一。该疾病并非单纯的“磨损”。瓣膜中的软性结缔组织细胞——称为瓣膜间质细胞——会转变为类似伤口修复或骨样的状态。它们沉积额外的胶原,然后沉积钙,使原本柔韧的瓣叶变成僵硬、岩石般的片状。作者分析了人类瓣膜组织和大型基因表达数据集,发现围绕小分子cGMP及其伴侣酶蛋白激酶G(PKG)构建的信号通路在钙化瓣膜中持续被下调,而与成骨相关的基因和蛋白被上调。严重瓣膜钙化患者血液中的cGMP水平也较低,并与瓣膜狭窄和阻塞程度相关。
失落的保护信号
为探究因果关系,研究团队使用了小鼠模型和培养的人瓣膜细胞。被设计为产生较少PKG的小鼠在对主动脉瓣进行受控损伤后,瓣叶更厚且钙化更重,其瓣膜中成骨主控蛋白水平更高。在鼓励钙沉积的人瓣膜细胞培养条件下,用遗传手段降低PKG会加速这种骨样转变。这些结果表明,cGMP‑PKG信号通常对瓣膜中有害的细胞重编程起制动作用——当这制动减弱时,钙化加速发生。
重新利用一种心衰药物
研究人员随后询问增强该通路是否能减缓或逆转钙化。他们测试了三种以不同方式增加cGMP的药物,其中包括已获批用于部分心衰患者的维立昔胺(vericiguat)。在人体瓣膜细胞中,三种药物均减少了钙沉积和成骨标志物,其中维立昔胺作用最强。维立昔胺也减少了体外维持活性的微小人瓣膜组织的钙化。在两种小鼠模型中——一种由高胆固醇饮食驱动,另一种由机械损伤驱动——每日给予维立昔胺使瓣叶更薄、钙化更少且瓣膜跨流量改善,同时不削弱心脏的泵血功能。然而,当PKG被基因下调时,维立昔胺的大部分益处消失,表明PKG是关键的下游效应物。
细胞清理,瓣膜更健康
进一步探查时,团队发现维立昔胺与PKG保护瓣膜细胞的线粒体。在促钙条件下,细胞积累有害的活性氧,线粒体膜电位丧失,能量产出减少。维立昔胺恢复了线粒体功能并减少氧化应激。大规模蛋白质与磷酸化位点谱图指向自噬——细胞的内部清理与回收系统。在钙化的人瓣膜中,显微成像和蛋白标志显示自噬体(自噬的“垃圾袋”)的形成与活性降低。单细胞分析显示,病变瓣膜的若干细胞亚型广泛抑制自噬相关程序。在培养细胞中,阻断自噬会抹去PKG激活的保护效应,暗示PKG主要通过恢复这一清理系统发挥作用。
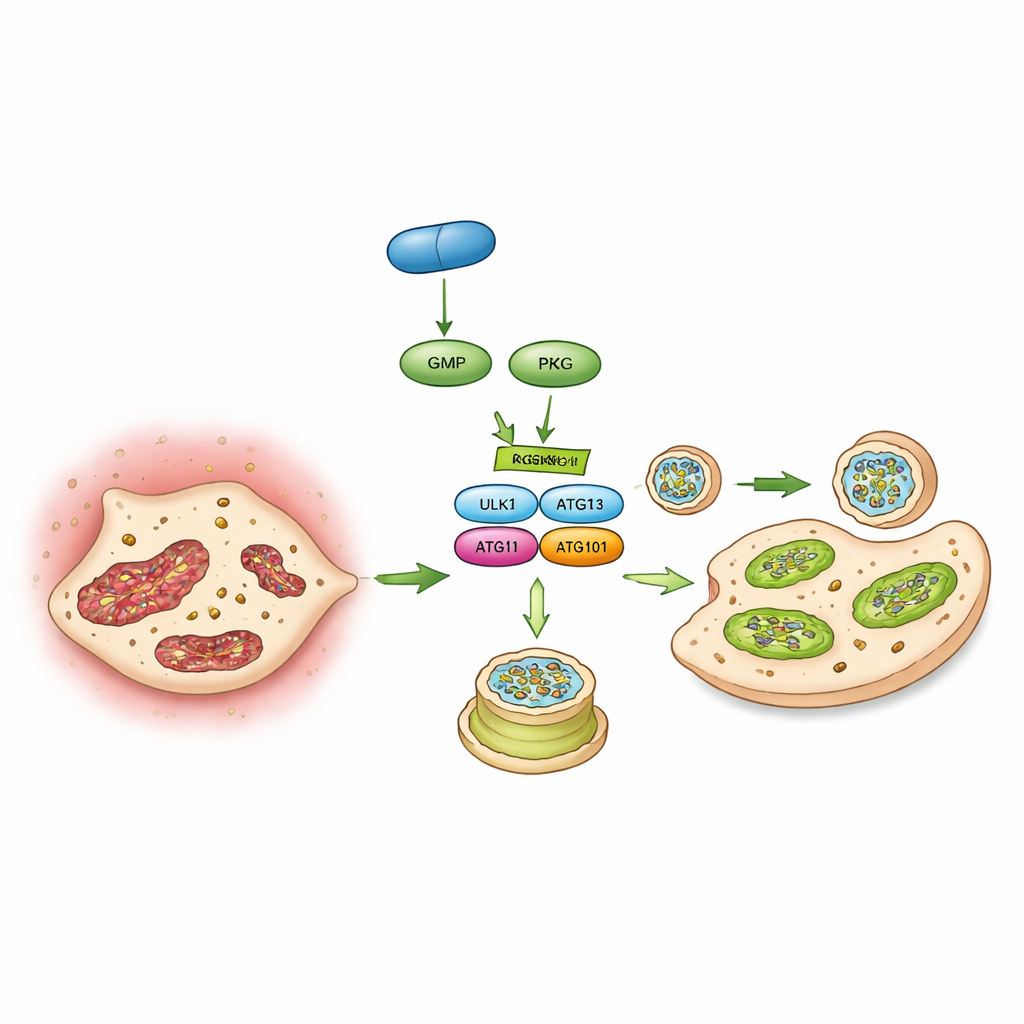
翻转一个分子开关
最后,作者确定了将PKG与自噬连接起来的特异分子开关:一种名为ULK1的蛋白,它启动自噬体的形成。通过磷酸化蛋白组学、相互作用研究和试管激酶实验,他们证明PKG直接在ULK1的特定位点(称为丝氨酸556)上加磷酸。当该位点被突变以阻止修饰时,PKG无法增强自噬或阻断瓣膜细胞的钙化。在小鼠中,在瓣膜细胞中驱动表达这种对PKG无反应的ULK1突变体也消除了维立昔胺的保护作用。综合这些发现勾勒出一条事件链:维立昔胺刺激cGMP,激活PKG,PKG翻动ULK1开关,复苏自噬,保护线粒体并阻止瓣膜细胞向成骨细胞转变。
这对患者可能意味着什么
这项工作将cGMP–PKG–ULK1–自噬轴定位为对抗主动脉瓣“生锈”的内在防御系统。在钙化性瓣膜疾病中,该防御受损,使细胞积累损伤并向骨样身份转变。通过药理学手段恢复这一信号——使用已在心衰临床中应用的药物——研究者在多种实验模型中减缓了钙化。尽管仍需在人类瓣膜疾病中进行临床试验,但该研究提出了一个清晰且可检验的设想:谨慎增强这一细胞清理通路,或许有一天能推迟或减少老年主动脉瓣狭窄高危患者的瓣膜置换手术需求。
引用: Wang, Y., Xu, F., Song, C. et al. Cyclic guanosine monophosphate-protein kinase G signaling attenuates aortic valve calcification through ULK1-mediated autophagy. Sig Transduct Target Ther 11, 90 (2026). https://doi.org/10.1038/s41392-026-02624-5
关键词: 钙化性主动脉瓣疾病, cGMP PKG 信号, 维立昔胺(vericiguat), 自噬, 心脏瓣膜钙化