Clear Sky Science · tr
Oda ve üzerindeki sıcaklıklarda benzersiz dayanıklılık gösteren fizik bilgili atomik enerji modelleri
Günlük kimya için neden önemli
Bilgisayar simülasyonları modern kimya ve malzeme biliminin iş atlarıdır. Bilim insanlarının pahalı ve zaman alıcı deneyler yerine moleküllerin dönmesini, titreşmesini ve çarpışmasını bilgisayar ortamında izlemesine olanak tanırlar. Ancak bu simülasyonlar makine öğrenmesine dayandığında, özellikle daha yüksek sıcaklıklarda ansızın "çöker" ve imkansız moleküler şekiller üretebilirler. Bu çalışma, böyle simülasyonları son derece uzun süreler boyunca, 1000 kelvine kadar sıcaklıklarda çalıştırabilen ve dağılmayan yeni bir fizik bilgili makine öğrenmesi modelini tanıtıyor.
Akıllı kestirmelerden kırılgan simülasyonlara
Geleneksel kuantum kimyası moleküler enerjileri yüksek doğrulukla hesaplar fakat son derece yavaştır. Daha basit kuvvet alanları hızlıdır ama genellikle yaklaşık çözümler sunar. Makine öğrenimli potansiyeller her iki dünyanın en iyisini birleştirmeyi hedefler: moleküler geometriden enerji ve kuvvetlere bir kestirme öğrenir ve bu kestirmeyi moleküler dinamikleri sürdürmek için kullanır. Kağıt üzerinde birçok böyle model mükemmel görünür; standart test setlerindeki ortalama hatalar çok küçüktür. Uygulamada ise bu sayılar aldatıcı olabilir. Moleküller bir simülasyon sırasında—özellikle yüksek sıcaklıklarda—yeni şekiller keşfettiğinde, birçok model eğitim gördüğü yapı aralığının dışına itilir. Molekülleri gerçekçi şekillere nazikçe yönlendirmek yerine bağları gerebilecek veya ezebilecek kuvvetler tahmin edebilirler; bu da sistemin fizik dışı hale gelmesine ve simülasyonun çökmesine yol açar.
Kuantuma dayalı yapı taşları üzerine model kurma
Yazarlar bu kırılganlığı, modelin neyi öğrendiğini ve önceden tanımlanmış fizik bilgileriyle nasıl yönlendirildiğini değiştirerek ele alıyor. FFLUX adında, Interacting Quantum Atoms (IQA) yaklaşımına dayanan bir çerçeve kullanıyorlar. IQA’da bir molekül, bireysel enerjileri doğrudan kuantum mekaniğinden belirlenen "topolojik atomlara" bölünür. Bu atomik enerjiler fiziksel olarak anlamlıdır ve molekülün toplam enerjisine toplanır. Rastgele site enerjileri öğrenmek yerine yeni Gauss süreçli modeller bu kuantum kökenli atomik enerjileri öğrenir; bu da her tahmin için derin bir fiziksel çapa sağlar. Dört esnek organik molekül—peptid kaplı glisin ve serin, malondialdehit ve aspirin—iç hareketleri çok olduğundan ve mevcut makine öğrenmeli kuvvet alanları için bilinen zorluklar taşıdığından dolayı zorlu test yatakları olarak kullanıldı.
Modele sorun beklemeyi öğretmek
Temel yenilik, Gauss sürecinin veriyi hiç görmeden önce nasıl kurulduğunda yatıyor: modelin az bilinen bölgelerde ne beklediğini kodlayan "ortalama fonksiyonu". Önceki çoğu çalışmada bu ortalama basitçe sıfıra ayarlanır; bu da modelin ön bilgiye sahip olmadığını varsaymak gibidir. Yazarlar bunun yerine bu ortalamayı, fiziksel olarak mantıklı kalacak şekilde, daha yüksek atomik enerji durumlarına doğru kasıtlı olarak kaydırıyorlar. Bu tasarım seçimi, modelin dışa taşma yapmak zorunda kaldığında—örneğin bağlar geçici olarak aşırı uzadığında—aşırı bozulmaları cezalandıran tahminleri doğal olarak tercih etmesini sağlıyor. Geniş testlerde, yalnızca bu ön bilgi açısından farklı olan model versiyonları çok farklı davrandı. Geleneksel veya düşük enerjili ortalamalara sahip modeller genellikle bir molekül patlamadan veya çökmeye uğramadan önce bir pikosaniyeden az süre hayatta kaldı. Buna karşın en iyi yüksek enerjili ortalama (MF5 adı verilen) tüm dört molekül için 300 ile 1000 kelvin arasındaki sıcaklıklarda tam nanosaneye kadar süren test penceresinde kararlı simülasyonlar verdi.
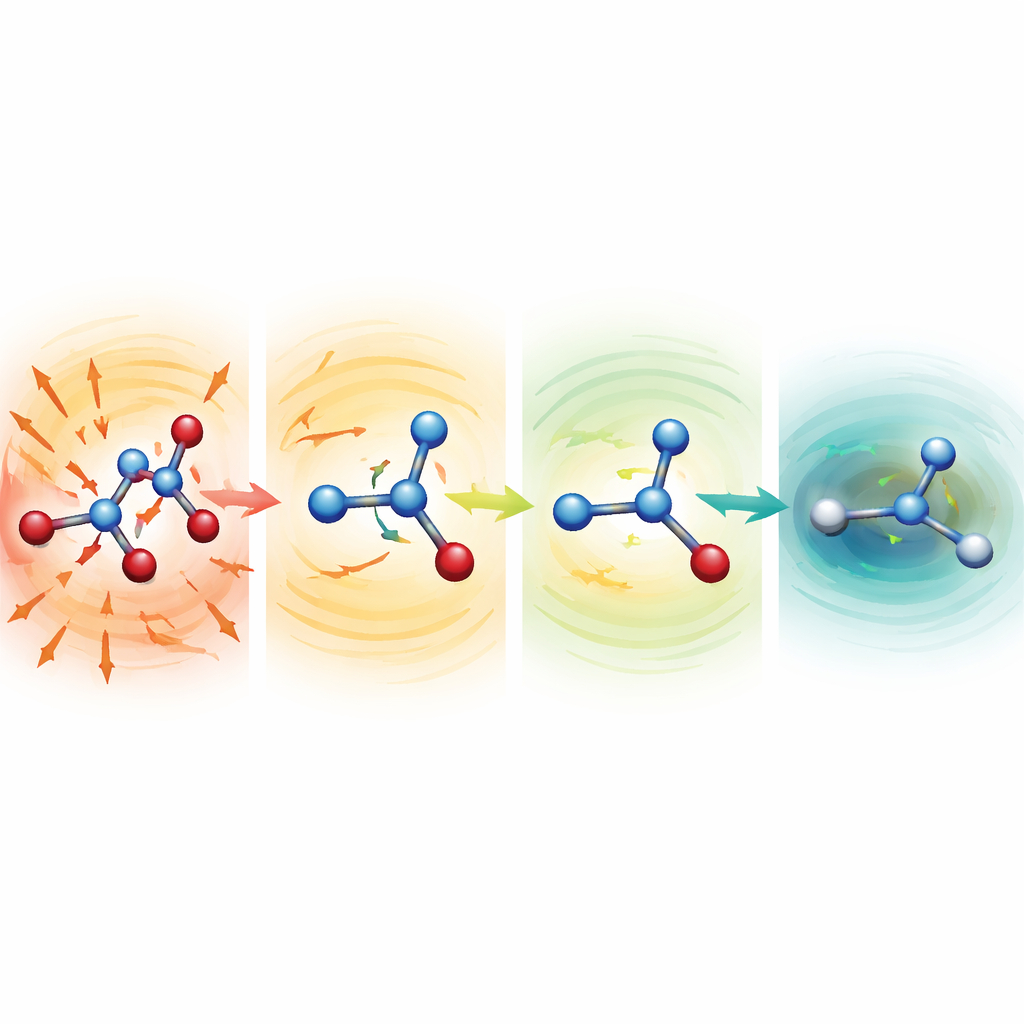
Bozulmuş moleküllerin kendini onarmasını izlemek
Dayanıklı modellerin neden bu kadar iyi çalıştığını araştırmak için araştırmacılar, simülasyonları kasıtlı olarak ciddi şekilde gerilmiş veya sıkışmış bağlara sahip bozulan yapılardan başlattı. Serin, aspirin ve malondialdehit için bu başlangıç noktaları normal bir yapının yüzlerce ila binlerce kilokalori/ mol üzerinde yer alıyordu—normalde felaketle sonuçlanacak konfigürasyonlar. Zayıf ortalama fonksiyonlarla moleküller hızla dağıldı. Ancak MF5 düzeninde tahmin edilen kuvvetler anında uzunlamasına bağları kısaltan ve ezilmiş olanları uzatan yönleri işaret etti. Onlarca ila yüzlerce zaman adımı içinde moleküller gerçekçi şekillere gevşedi ve sonra kararlı bir şekilde evrimleşmeye devam etti. Ekip ayrıca, kuvvetler üzerinde hiç eğitilmemiş olmalarına rağmen aynı modellerin alanin dipeptidin geometri optimizasyonlarını yönlendirebildiğini gösterdi; bilinen düşük enerjili konformasyonları ve göreli enerjileri birkaç ondalık kilokalori/ mol içinde yeniden ürettiler, ancak tam kuantum hesaplarına kıyasla yaklaşık 200 kat daha düşük maliyetle.
Günlük donanımda uzun, sıcak simülasyonlar
Dayanıklılık sadece tek bir zor başlangıca dayanmamak; milyonlarca veya milyarlarca zaman adımı boyunca koruyabilmekle ilgilidir. Yazarlar en iyi modellerini daha ileri zorlayarak, dört test molekülleri için her biri 10 nanosecond süren 500 kelvin sıcaklıkta 50 bağımsız simülasyon yürüttü. Bu koşu boyunca hiçbir çalışma çökmedi ve toplam yarım mikrosoniye ulaşan birleşik simülasyon süresi elde edildi—bu, son teknoloji makine öğrenmeli kuvvet alanları için olağan dışı bir sonuç. Daha da çarpıcı şekilde, simülasyonlar standart CPU’larda verimli biçimde çalıştı; adım adım bazı önde gelen GPU gerektiren sinir ağı potansiyelleriyle boy ölçüşüyor ya da onları geçiyordu. Simülasyonlar boyunca moleküller zengin şekil ve meta-kararlı durum setlerini keşfetti; bu da dayanıklılığın hareketi yapay biçimde dondurarak veya katı yapılar uygulayarak elde edilmediğini gösteriyor.
Geleceğin moleküler modellemesi için ne anlama geliyor
Uzman olmayanlar için ana mesaj, düşük hatalı tüm makine öğrenmeli modellerin zorlandığında güvenilir olmadığıdır. Modellerini kuantum türevi atomik enerjilerle temellendirip modelin yerleşik beklentilerini yüksek enerji durumlarına doğru dikkatle ayarlayarak, yazarlar doğal olarak "iyileştirici kuvvetler" üreten—simülasyonları yüksek sıcaklıklarda ve bozulmuş başlangıç noktalarında bile fiziksel tutan—bir potansiyel ailesi yarattı. Bu yaklaşım karmaşık moleküllerin daha güvenilir ve daha uzun simülasyonlarını vaat ediyor ve benzer fizik bilgili modellerin yoğun fazları ve dağılım gibi ince etkileşimleri ele alabileceği gelecekteki uzantılara işaret ediyor; tüm bunlar hesaplama açısından pratik kalırken mümkün olacak.
Atıf: Isamura, B.K., Aten, O., Nosratjoo, M. et al. Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature. Commun Chem 9, 138 (2026). https://doi.org/10.1038/s42004-026-01965-0
Anahtar kelimeler: makine öğrenmeli kuvvet alanları, moleküler dinamiklerin dayanıklılığı, Gauss süreç potansiyelleri, fizik bilgili modelleme, kuantum atomik enerjileri