Clear Sky Science · tr
Pakistan’da akraba evlilikleri olan ailelerde resesif kalıtımlı ataksik ve nöropatik bozuklukların moleküler karakterizasyonu
Aileler ve hekimler için neden önemli
Denge, yürüyüş ve el-ayaklardaki duyu sorunları özellikle çocuklukta başlayıp yaşam boyu yavaşça kötüleştiğinde son derece sakatlayıcı olabilir. Kuzenlerin sık evlendiği bölgelerde bu semptomlar pek çok ailede birkaç kuşak boyunca açıklanmadan devam edebilir. Bu çalışma, Pakistan’daki bu tür aileler için acil bir soruyu ele alıyor: modern DNA analizleri, ataksi (zayıf denge ve koordinasyon) ve periferik nöropatinin (uzuvlardaki sinir hasarı) arkasındaki gizli genetik nedenleri nihayet ortaya çıkarıp hekimlerin daha net tanı ve olası tedavi seçenekleri sunmasına yardımcı olabilir mi?
Büyük ailelerde kalıtılan hastalığı izlemek
Araştırmacılar, birkaç üyesinde ciddi hareket ve sinir sorunları olan yedi geniş Pakistanlı aile ile çalıştı. Bazı kişilerde ağırlıklı olarak ataksi vardı; bu durum sabit yürüme ve konuşma ile göz hareketlerinin kontrolünü zorlaştırıyordu. Diğerlerinde ise ellerde ve ayaklarda kas erimesi, ayak şekil bozuklukları ve refleks kaybı gibi klasik periferik nöropati belirtileri görüldü. Bu ailelerde ebeveynler birbirleriyle akrabaydı ve bu da çocukların aynı nadir kusurlu genin iki kopyasını miras alma olasılığını artırıyordu. Etkilenen ve etkilenmeyen akrabalardan alınan kan örnekleri kullanılarak ekip, hastalığın aile ağaçları boyunca izlediği zararlı değişiklikleri bulmak amacıyla eksom dizilemesi—genomun protein kodlayan bölümlerinin büyük kısmını okuma—yaptı. 
Nadir kusurlu genleri belirlemek
Yaygın ve zararsız DNA farklılıklarını süzdükten sonra bilim insanları yedi ailenin beşinde muhtemel hastalık etmenleri tespit etti. Bu ailelerin her biri kendi özgü genetik değişikliğini taşıyordu ve tümü resesif bir modele uyuyordu: bireyler yalnızca ebeveynlerden birer kusurlu kopya alarak iki kopyaya sahip olduklarında hasta oluyordu. Yetişkin başlangıçlı denge sorunları ve konuşma güçlüğü gösteren bir ailede, suçlu lizozom adı verilen hücre geri dönüşüm bölmelerine materyal taşıyan MFSD8 genindeki nadir bir değişiklikti. Bir diğer ailede, hücrenin enerji santralleri olan mitokondrilerin sağlığını koruyan bir protein olan AFG3L2’deki zararlı değişiklik çocukluk başlangıçlı spastik ataksiyle birlikte distoni (anormal kas kasılmaları) ile ilişkilendirildi. Üçüncü bir ailede ise DNA’yı onarım sırasında koruyan ve göz hareketlerini de etkileyen okülomotor apraxi ile ilişkili ataksiyi bilinen şekilde yapabilen SETX geninde bir çerçeve kayması (frameshift) hatası bulundu.
Kalıtsal sinir hasarına daha yakından bakış
İki başka ailede ise ayaklara ve ellere giden uzun sinirlere zarar veren kalıtsal durumlar grubundan Charcot‑Marie‑Tooth (CMT) hastalığının bir biçimi tespit edildi. Her iki ailede de araştırmacılar sinir hücrelerinde normal mitokondri işlevi için kritik olan GDAP1 geninde zararlı varyantlar buldu. Bir GDAP1 değişikliği proteini kısaltıyor ve çok şiddetli, erken başlangıçlı hastalıkla ilişkilendiriliyordu; diğer değişiklik ise proteindeki tek bir yapı taşını değiştiriyor ve daha ılımlı bir seyirle ilişkiliydi. Çarpıcı bir şekilde, bir CMT ailesindeki en ağır etkilenen hasta ayrıca vitamin B12 işlenmesinde rol oynayan ve bazen vitamin temelli tedavilerle düzeltilebilen MMACHC geninde bilinen bir hastalık varyantı için homozigot durumdaydı. Bu çift etkili durum, semptomlarının MMACHC varyantını taşımayan akrabalarından daha kötü olmasının nedenini açıklayabilir.
DNA araması yeterli olmadığında
Her aile net bir genetik cevaba ulaşamadı. Yedi ailenin ikisinde ekip, eksomda hastalık paterniyle ikna edici şekilde eşleşen tek bir değişiklik bulamadı. Bir vakada, kalıtım modeline uyan EHHADH geninde bir varyant tespit ettiler ancak bu değişikliğin zararsız olması öngörülüyor ve değiştiğinde farklı bir böbrekle ilişkili duruma yol açtığı biliniyor. Başka bir durumda ise benzer hareket sorunları olan iki kuzenin farklı altta yatan nedenleri olduğu ortaya çıktı: bir erkek bilinen zararlı bir ALS2 varyantını taşıyordu ki bu genç yaşta motor nöron hastalıklarına yol açabiliyor; oysa etkilenen kuzenleri bu varyantı taşımıyordu. Bu çözülemeyen vakalar, önemli mutasyonların standart eksom dizilemesinin yakalayamadığı genom bölgelerinde olabileceğini veya birden fazla ince genetik faktörün etkileşiyor olabileceğini düşündürüyor.
Hastalar ve gelecekteki bakım için ne anlama geliyor
Sonuçlar birlikte güçlü DNA araçlarının, sınırlı kaynakların bulunduğu ortamlarda bile karmaşık sinir ve denge bozukluklarının arkasındaki spesifik genleri ortaya çıkarabileceğini gösteriyor. Net bulgular saptanan beş aile için çalışma, “ataksi” veya “nöropati” gibi belirsiz etiketleri belirli genlerle ilişkilendirilen kesin tanılara dönüştürüyor; bu da genetik danışmanlığa rehberlik edebilir, prognozu bilgilendirebilir ve bazı durumlarda MMACHC ile ilişkili hastalıklar için vitamin B12‑ile ilgili tedavi gibi tedavi seçeneklerini öne çıkarabilir. Çalışma ayrıca MFSD8, AFG3L2, SETX, GDAP1, MMACHC ve ALS2 gibi genlerin beyin, omurilik ve periferik sinirler genelindeki sinir hücresi sağlığını nasıl şekillendirdiğine dair bilim insanlarının anlayışını genişletiyor. İleriye bakıldığında, kalan gizemleri çözmek ve bu genetik bilgileri etkilenen çocuklar ve yetişkinler için daha erken tanı ve daha iyi bakıma çevirmek üzere daha kapsamlı genom dizilemesi ve fonksiyonel çalışmalar gerekecektir. 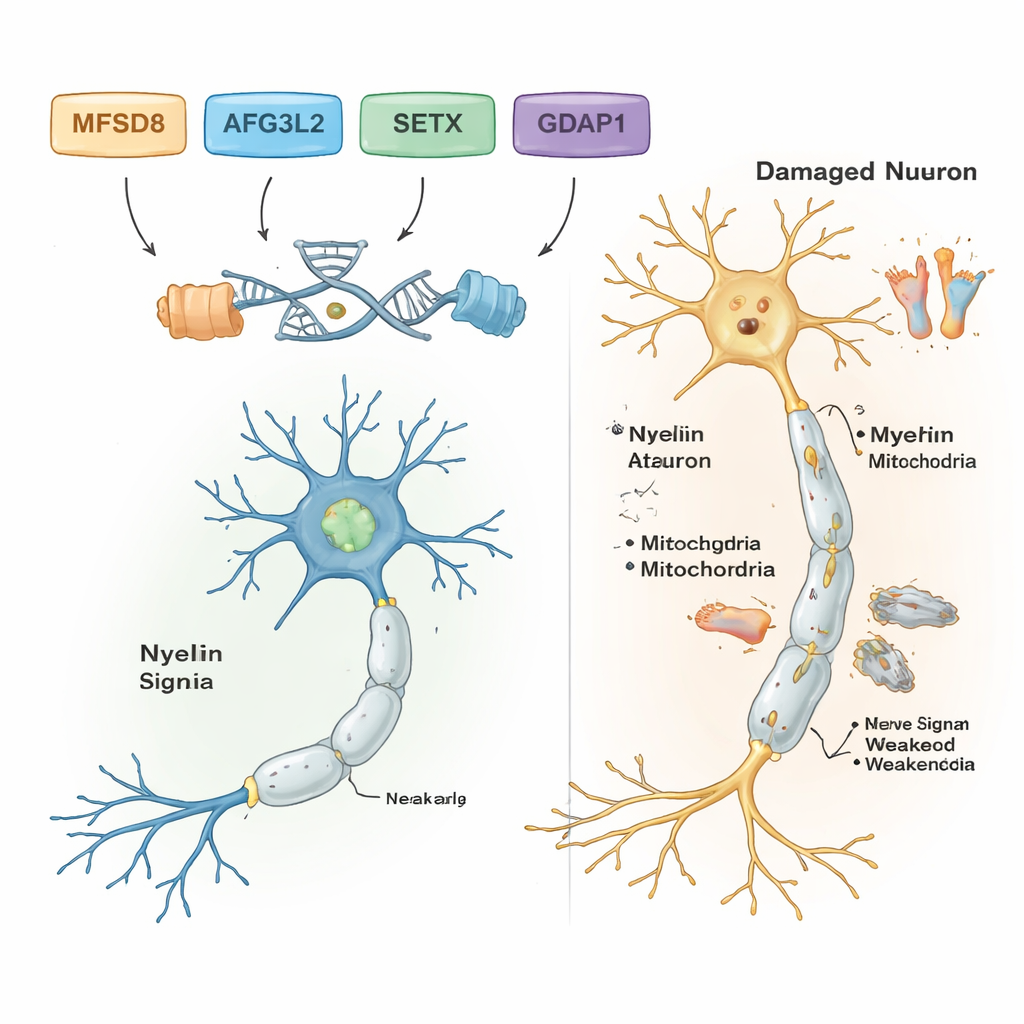
Atıf: Aslam, F., Wajid, M., Butt, A.I. et al. Molecular characterization of recessively inherited ataxic and neuropathic disorders in consanguineous Pakistani families. Sci Rep 16, 6529 (2026). https://doi.org/10.1038/s41598-026-37808-0
Anahtar kelimeler: ataksi, periferik nöropati, eksom dizilemesi, Charcot-Marie-Tooth hastalığı, genetik tanı