Clear Sky Science · tr
ATP13A2 ekspresyonunun baskılanmasıyla ilişkili mitokondriyal disfonksiyon yoluyla hücre içi demir dengesinin bozulması
Beyin hücreleri içindeki demirin önemi
Parkinson hastalığı titreme ve sert hareketlerle tanınsa da, etkilenen beyin hücrelerinin derinlerinde başka bir drama gerçekleşir: hayati bir metal olan demir, olmaması gereken yerlerde birikmeye başlar. Bu çalışma basit ama önemli bir soruyu soruyor: bu demir birikimi nasıl oluşuyor ve sinir hücrelerinin içindeki küçük enerji santralleri ile geri dönüşüm merkezlerine nasıl zarar verebilir? Buna yanıt arayarak çalışma, Parkinson ve ilgili bozukluklarda belirli beyin bölgelerinin neden dejenerasyona uğradığına dair ipuçları sunuyor ve dopamin yerine koyma tedavilerinin ötesine geçen yeni tedavi yollarına işaret ediyor.
Nadir bir genetik ipucuna daha yakından bakış
Araştırmacılar, ATP13A2 adlı gendeki kusurlardan kaynaklanan nadir kalıtsal Parkinson formu PARK9 üzerinde odaklanıyor. Bu gen, hücrenin atık işleme ve geri dönüşüm bölmeleri olan lizozomlarda bulunan bir protein üretir. ATP13A2 mutasyonları olan kişilerde beyinde demir birikimi ile karakterize bir tablo da gelişebilir. Bu bağlantı ATP13A2'yi demir dengesinin nasıl bozulduğunu incelemek için ideal bir başlangıç noktası yaptı. Parkinson proteini alfa-sinükleinini aşırı üreten insan nöron benzeri bir hücre hattı kullanılarak, ekip küçük RNA parçaları ile ATP13A2'yi susturdu ve ardından demir, enerji üretimi ve hücre sağlığının nasıl değiştiğini izledi.
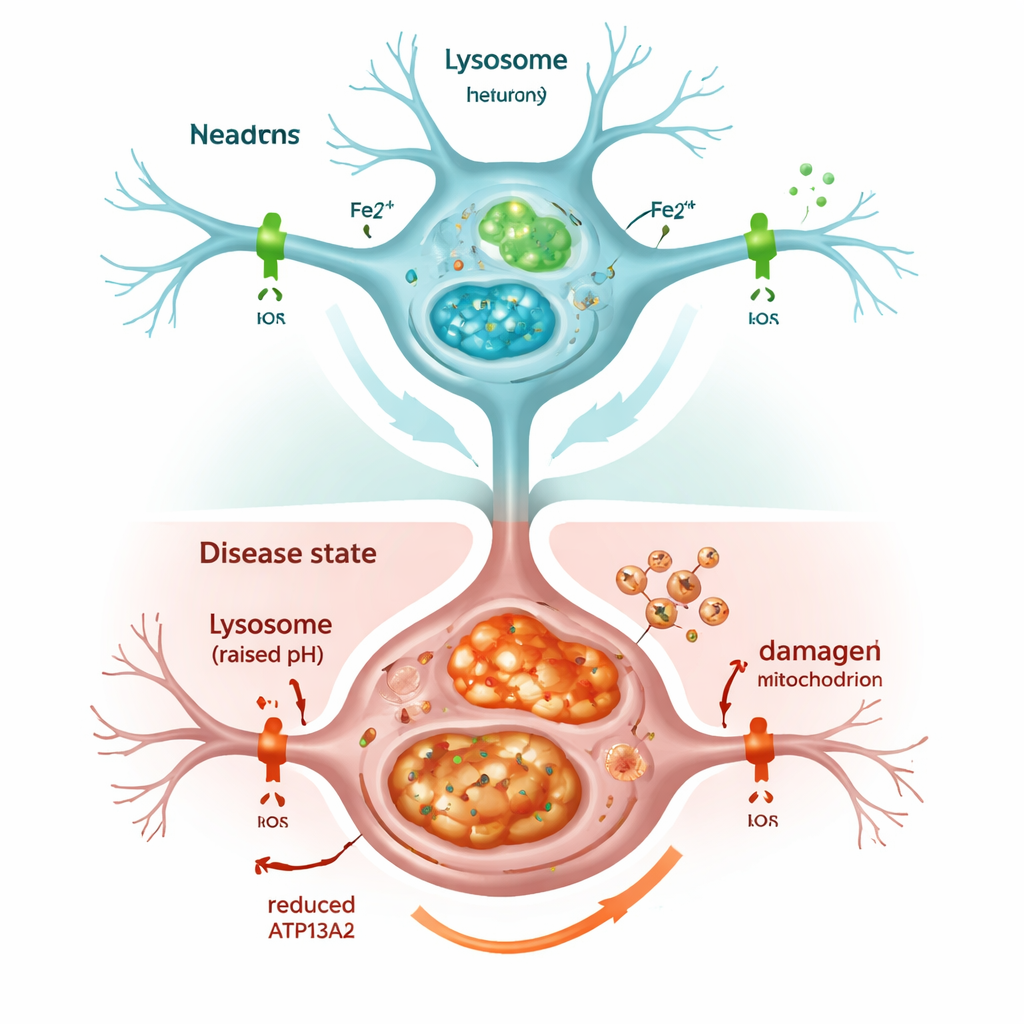
Hücrenin geri dönüşüm sistemi durduğunda
ATP13A2'nin kapatılması lizozomları hızla zayıflattı. İstenmeyen maddeleri parçalamak için kritik olan iç asitlik azaldı ve otofaji olarak bilinen hücresel temizlik sürecinin göstergeleri temizlenmek yerine birikti. Sonuç olarak alfa-sinüklein birikti; bu, Parkinson beyinlerinde görülen duruma benziyordu. Hücreler ayrıca genel olarak daha fazla demir ve özellikle lizozomlarda ve mitokondrilerde kimyasal olarak aktif form olan Fe2+ artışı gösterdi. Hücre, demiri depolayan ferritin adlı proteini daha fazla üreterek yanıt verdi, ancak bu sorunların önüne geçmeye yetmedi: aşırı yüklü mitokondriler fazla reaktif oksijen türleri üretti ve hücre canlılığı düştü. Hücrelere bazı kliniklerde kullanılanlara benzer bir demir bağlayıcı ilaç verildiğinde oksidatif stres azaldı ve hücre yaşama kısmen geri döndü; bu da fazla demirin hasarın temel sürücüleri arasında olduğunu vurguladı.
Demir algılayıcıları metali dinlemeyi bırakıyor
Normalde hücrelerin demir seviyeleri yükseldiğinde bunu algılayıp demir girişini azaltan bir geri besleme sistemi vardır. IRP2 adlı bir protein, kısmen mitokondrilerden gelen heme bağımlı bir sinyal aracılığıyla demiri algılar ve ardından hücre yüzeyindeki demir taşıyıcı proteinlerin üretimini ayarlar. ATP13A2 eksik hücrelerde bu koruma mekanizması başarısız oldu. Hücreye demir getiren taşıyıcılar, demir zaten yüksek olmasına rağmen yüksek kaldı. IRP2 protein düzeyleri neredeyse değişmedi ve dışarıdan ekstra demir eklemek normalde yol açtığı yıkımı tetiklemedi. Ekip bu başarısızlığı mitokondrilere bağladı: hasarlı mitokondriler daha az verimli solunum yaptı, mitofaji (mitokondri kalitesini koruma) kusurlarına işaret etti ve en önemlisi heme yapma yeteneğini kaybetti; heme IRP2'nin demiri algılamasına yardımcı olan demir içeren moleküldü. Yeterince heme olmadığında IRP2 "çok fazla demir" mesajını alamadı ve sürekli demir girişine izin verdi.
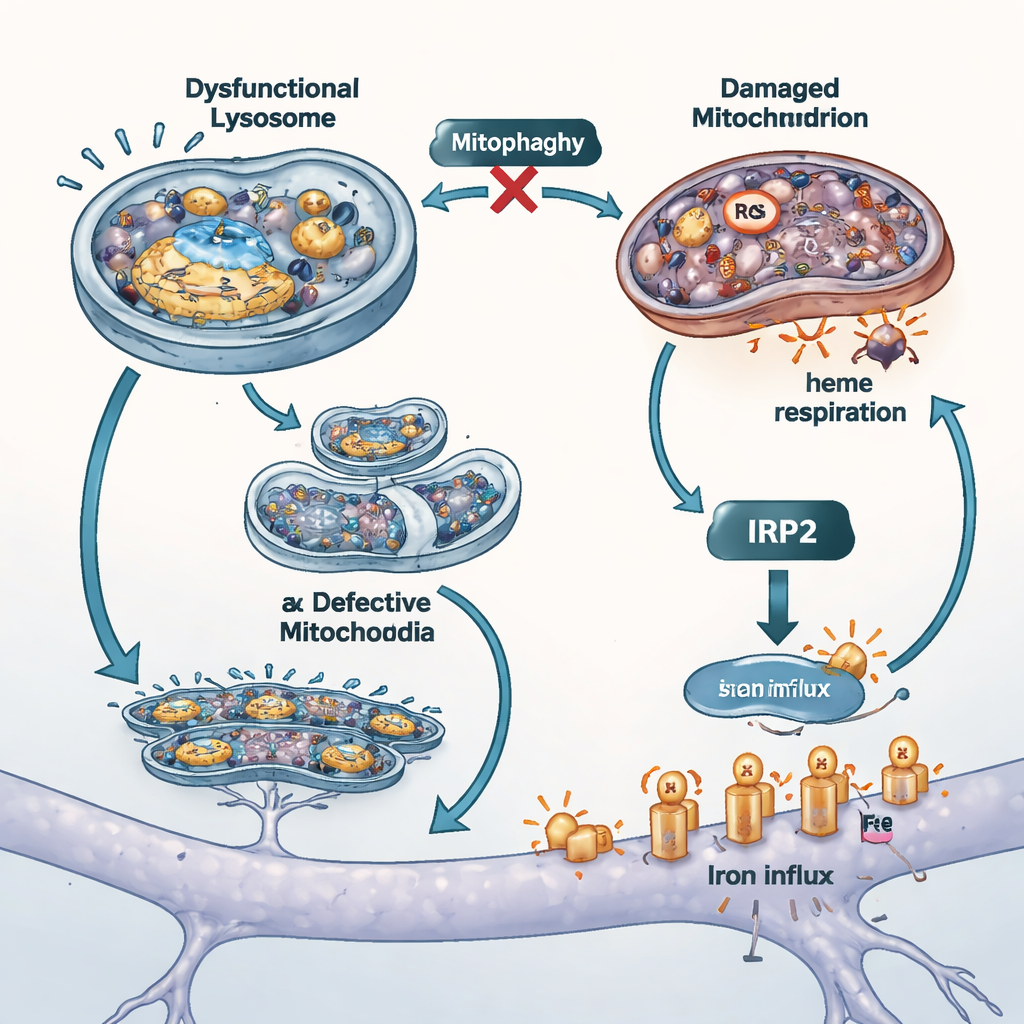
Demir musluğunu kapatmak ve diğer modelleri test etmek
Bilim insanları bu kontrolsüz demir girişinin hücre hasarına ne kadar katkıda bulunduğunu araştırmak için iki ana demir alım yolunu bloke etti. Bir alıcıyı rekabetle dışlamak için demirsiz bir transferrin versiyonu kullandılar ve DMT1 adlı başka bir taşıyıcının aktivitesini azaltmak için küçük bir ilaç kullandılar. Her iki müdahale de hücre içindeki toplam ve serbest demiri azalttı, mitokondriyal oksidatif stresi düşürdü ve canlılığı iyileştirdi; bu, ATP13A2 kaybında yüzey demir kanallarının hasarı güçlendiren önemli amplifikatörler olduğunu gösterdi. Araştırmacılar ayrıca mitofajiyi bozduğu bilinen başka bir Parkinson bağlantılı gen olan PINK1 eksik hücrelerde kilit deneyleri tekrarladı. Bu hücreler aynı demir birikimi ve zayıflamış heme üretimi kombinasyonunu gösterdi; bu da mitokondriyal kalite kontrol ile demir dengesinin hastalığın farklı formlarında sıkı şekilde birbirine bağlı olduğunu destekliyor.
Parkinson ve gelecekteki tedaviler için anlamı
Basitçe ifade etmek gerekirse, çalışma bir kısır döngüyü özetliyor. ATP13A2 baskılandığında lizozomlar hasarlı bileşenleri, aralarında kusurlu mitokondrileri temizleyemez. Bu zayıflamış mitokondriler daha az enerji ve daha az heme üretir ve hücrenin demir algılama sistemini köreltir. Demir yüzey taşıyıcıları aracılığıyla akmaya devam eder, hassas bölümlerde birikir ve mitokondrilere daha fazla zarar veren toksik reaksiyonları körükler. Zaman içinde bu döngü, belirli nöronların Parkinson’da ve ilgili demir yüklenmeli beyin bozukluklarında neden öldüğünü açıklamaya yardımcı olabilir. Bulgular, gelecekteki tedavilerin yalnızca fazla demiri uzaklaştırmayı değil, aynı zamanda uygun lizozom fonksiyonunu, mitokondriyal kalite kontrolü ve heme üretimini de onarmayı hedefleyebileceğini — sorunu kaynağında ele almanın, metalin sonradan temizlenmesinden daha etkili olabileceğini öne sürüyor.
Atıf: Murakami, T., Ohuchi, K., Kiuchi, M. et al. Disruption of intracellular iron homeostasis through mitochondrial dysfunction associated with suppression of ATP 13A2 expression. Sci Rep 16, 5007 (2026). https://doi.org/10.1038/s41598-026-35368-x
Anahtar kelimeler: Parkinson hastalığı, beyin demiri, mitokondriler, lizozomlar, heme sentezi