Clear Sky Science · tr
AF2BIND: AlphaFold2’nin çift temsili kullanılarak küçük-molekül bağlanma bölgelerinin tahmini
Binlerce Protein Arasında İlaç Hedefleri Bulmak
Modern ilaçların çoğu, hücrelerimizdeki proteinlerin yüzeylerindeki küçük girinti ve çıkıntılara tutunarak etki eder. Ancak günümüzün geniş protein yapı kataloglarına rağmen, bir küçük molekülün—potansiyel bir ilacın—nereye gerçekten yapışacağını önceden kestirmek hâlâ şaşırtıcı derecede zordur. Bu çalışma, AlphaFold2’nin (öncü protein yapı tahmincisi) içsel verilerini kullanarak binlerce insan proteini arasında olası ilaç-bağlanma bölgelerini öne çıkaran basit ama güçlü hesaplamalı bir araç olan AF2BIND’i tanıtıyor. Amaç, yeni ilaç arayışını daraltmak ve geleneksel yöntemlerin gözden kaçırdığı gizli işlevsel sıcak noktaları ortaya çıkarmak.
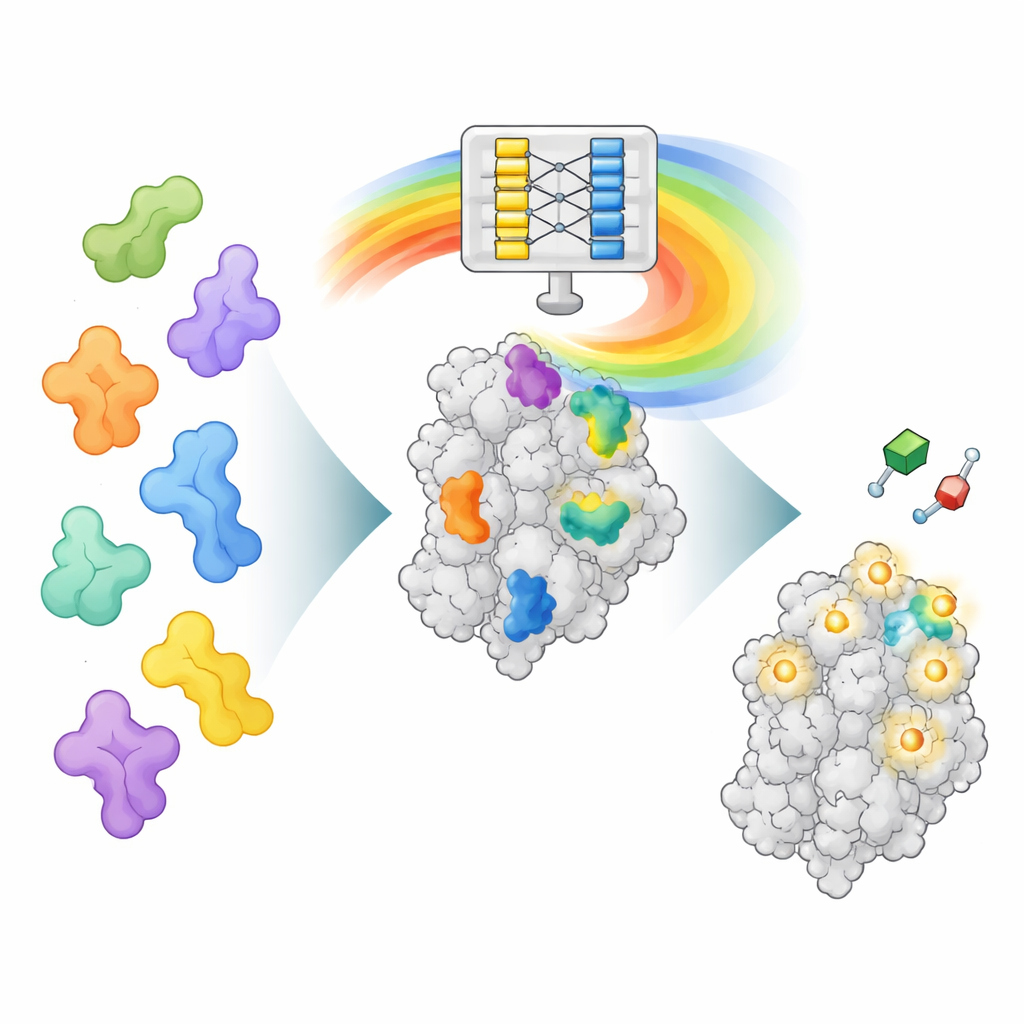
AlphaFold’un “Zihnini” Okumanın Yeni Bir Yolu
AlphaFold2, bir amino asit dizisinin üç boyutlu bir proteine nasıl katlanacağını tahmin edecek şekilde eğitildi; ilaçların nerelere bağlanacağını bulmak için değil. Ancak proteinleri katlamayı öğrenirken, farklı protein parçalarının nasıl etkileştiğine dair zengin desenler de öğrendi. AF2BIND bu içsel veri katmanlarından biri olan ve her bir amino asit pozisyonu çiftinin mekânsal ilişkisini kodlayan çift temsilini kullanıyor. Yazarlar AlphaFold2’ye bir protein dizisini omurga yapısıyla birlikte veriyor ve ayrıca her bir türden olmak üzere 20 ekstra amino asidi ayrı “yem” zincirleri olarak ekliyorlar. AlphaFold2 daha sonra proteinin her bir yem kalıntısıyla nasıl etkileştiğini hesaplıyor. Bu etkileşim desenleri, bir pozisyondaki her bir amino asidin küçük-molekül bağlanma bölgesine ait olma olasılığını tahmin eden çok basit bir lojistik regresyon modelinin girdisini oluşturuyor.
Gizli Sinyalleri Pratik Tahminlere Dönüştürmek
AF2BIND’in eğitimi, küçük moleküllerin yüksek kaliteli deneysel kanıtlarla bağlandığı yaklaşık 1.900 protein–ligand yapısından dikkatle seçilmiş bir veri kümesi gerektirdi. Araştırmacılar, benzerlik yoluyla “kopya çekmeyi” önlemek için büyük çaba gösterdiler: verilerini öyle böldüler ki test proteinleri eğitimde kullanılanlarla genel kat, dizi veya hatta bağlanma cebi şekli açısından paylaşım göstermiyordu. Bu titiz kıyaslamada, AF2 çift temsili, yalnızca diziye dayanan veya yapıya koşullu dizi tasarımına dayananlar da dahil olmak üzere birkaç alternatif sinir ağı gömme yöntemini geride bıraktı. Sadece çift özelliklerini kullanarak AF2BIND, en üst sıralanan tahminlerde bilinen bağlanma kalıntılarının yaklaşık üçte ikisini geri buldu ve standart sınıflandırma metriklerinde güçlü performans gösterirken, protein şekli ve yan zincir yönelimlerindeki ılımlı değişikliklere karşı da dayanıklı kaldı.
Yem Kalıntılarından Kimyasal İpuçları Okumak
AF2BIND basit bir lineer model olduğu için kararları modern bir yapay zeka sistemine göre alışılmadık derecede şeffaf. 20 yem aminoasidinin her biri, verilen bir protein pozisyonundaki nihai bağlanma skoruna ölçülebilir bir katkı yapıyor. Yaklaşık 2.000 protein–ligand kompleksindeki bu katkıları inceleyerek yazarlar, belirli yem kombinasyonlarının yağlı, karbon açısından zengin ligandlar için daha güçlü şekilde devreye girdiğini; diğerlerinin ise daha polar, suyla iyi etkileşen moleküller için öne çıktığını buldular. Başka bir deyişle, yem aktivasyon desenleri, bir cebin hangi tür küçük molekülleri tercih ettiğine dair kaba bir kimyasal parmak izi gibi davranıyor. Bu, gelecekte AF2BIND benzeri yaklaşımların yalnızca bir ilacın nerede bağlanabileceğini işaretlemekle kalmayıp aynı zamanda en uygun kimya türüne dair ipucu verebileceğini öne sürüyor.
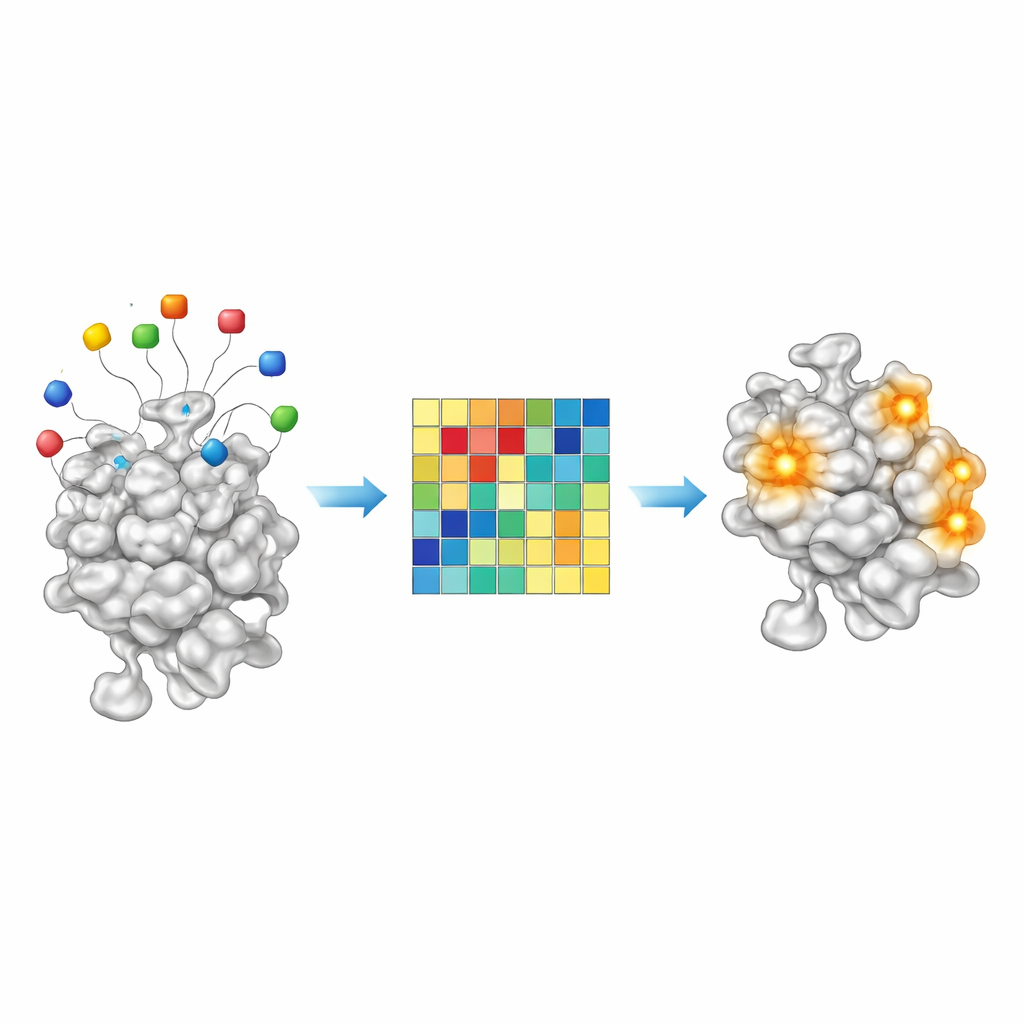
İnsan Proteomunu Yeni Cep'ler İçin Taramak
Eğitilmiş modelleriyle donanmış ekip, AF2BIND’i tüm insan proteomu için AlphaFold tarafından tahmin edilmiş yapılara uyguladı. Düşük güvene sahip bölgeleri kestikten ve çok büyük proteinleri yönetilebilir yapısal parçalara böldükten sonra, yakınlardaki yüksek puanlı kalıntıları aday bağlanma bölgelerine kümelendiler. AF2BIND, 13.000’den fazla proteinde 20.000’in üzerinde böyle bölge tahmin etti. Çarpıcı biçimde, bunların çoğu ligantları ilgili kristal yapılardan kopyalayan homoloji tabanlı yöntemler (ör. AlphaFill) veya yaygın kullanılan bir cep bulucu olan P2Rank ile örtüşmüyordu. AF2BIND’e özgü birçok bölge klasik gömülü ceplerden daha sığ veya daha yaygın yapıda olup sıklıkla peptid, RNA, DNA veya diğer proteinleri bağlayan bölgelerle—küçük moleküllerce hedeflenebilir olabilecek arayüzlerle—örtüşüyordu.
İlaç Keşfi ve Hastalık İçin Çıkarımlar
Bu yeni önerilen bölgelerin ilaç tasarımı açısından ne kadar umut verici olabileceğini değerlendirmek için yazarlar, cep boyutu, örtülme ve kimyasal çevre temelinde “ilaçlanabilirlik” puanı veren bağımsız bir aracı kullandılar. Ortalama olarak, AF2BIND’in bölgeleri, kalıplı ilaç hedefleri için yaygın bir eşik değerinin üstünde puan aldı; bunların arasında kalıtsal hastalıklarla ilişkilendirilen proteinlerde bulunanlar da vardı. Hücrelerdeki reaktif sisteinleri etiketleyen kemoproteomik deneylerle çapraz karşılaştırıldığında, AF2BIND ve P2Rank birlikte gözlemlenen ligandlanabilir bölgelerin neredeyse yarısını açıkladı; her iki yöntem de diğerinin kaçırdığı vakaları yakaladı. Bu çalışma, yapı-tahmin ağları tarafından öğrenilen içsel temsillerin, herhangi bir belirli ligand bilgisi olmadan olası ilaç-bağlanma bölgelerini büyük ölçekte haritalamak için yeniden kullanılabileceğini gösteriyor. Uzman olmayanlar için ana mesaj şu: protein şekillerini tahmin eden aynı yapay zeka atılımları, ilaçların bu şekilleri nerede ve nasıl kavrayabileceğini göstermeye başlıyor; bu da yeni tedavilerin bulunmasını hızlandırabilir ve proteinlerimizde daha önce gizli olan kontrol noktalarını aydınlatabilir.
Atıf: Gazizov, A., Lian, A., Goverde, C. et al. AF2BIND: predicting small-molecule binding sites using the pair representation of AlphaFold2. Nat Methods 23, 626–635 (2026). https://doi.org/10.1038/s41592-026-03011-2
Anahtar kelimeler: protein bağlanma bölgeleri, ilaç keşfi, AlphaFold2, hesaplamalı biyoloji, yapısal biyoinformatik