Clear Sky Science · tr
Pangenomun lineer referanslarını kullanarak eksik otizm varyantlarını keşfetmek
Gizli DNA değişiklikleri neden otizm için önemli
Genetik test yaptıran çoğu aile net yanıtlar umar, ancak yaklaşık beşte dördü kesin bir genetik açıklama alamaz. Bu çalışma, bunun başlıca nedenlerinden birine odaklanıyor: etkili birçok DNA değişikliği standart testlerin göremeyeceği kadar karmaşıktır. 51 otizmli aileden 189 kişinin neredeyse eksiksiz genomlarını oluşturarak ve bunları yeni, daha zengin bir “pangenom” referansıyla karşılaştırarak araştırmacılar, gelişmiş dizilemenin daha önce görünmez olan nadir mutasyonları ortaya çıkarabileceğini ve bunların bazı otizm ve ilişkili durumların açıklanmasına yardımcı olabileceğini gösteriyor.
Standart genetik testlerin ötesine bakmak
Geleneksel klinik testler, bir kişinin genomunu taramak için kısa DNA parçalarına dayanır. Bu yöntem birçok tek harflik değişiklik için iyi çalışır ama tekrarlı ya da yapısal olarak karmaşık bölgelerde—tam da bazı güçlü hastalık yapıcı mutasyonların gizlendiği yerlerde—başarısız olma eğilimindedir. Ekip, daha önce kısa-okuma genom, ekson veya gen paneli testlerinin otizm veya Rett-benzeri semptomlar için neden bulamadığı ailelere odaklandı. Çok daha uzun DNA bölümlerini okuyan uzun-okuma dizileme kullanarak 189 birey için yüksek kaliteli, fazlanmış (phased) genome montajları oluşturuldu. Bu, her kişinin ebeveynlerinden aldığı her kromozomun iki kopyasını çok az boşlukla yeniden inşa edebildikleri anlamına geliyor.
Yapısal varyantlar: büyük değişikliklerin büyük etkileri
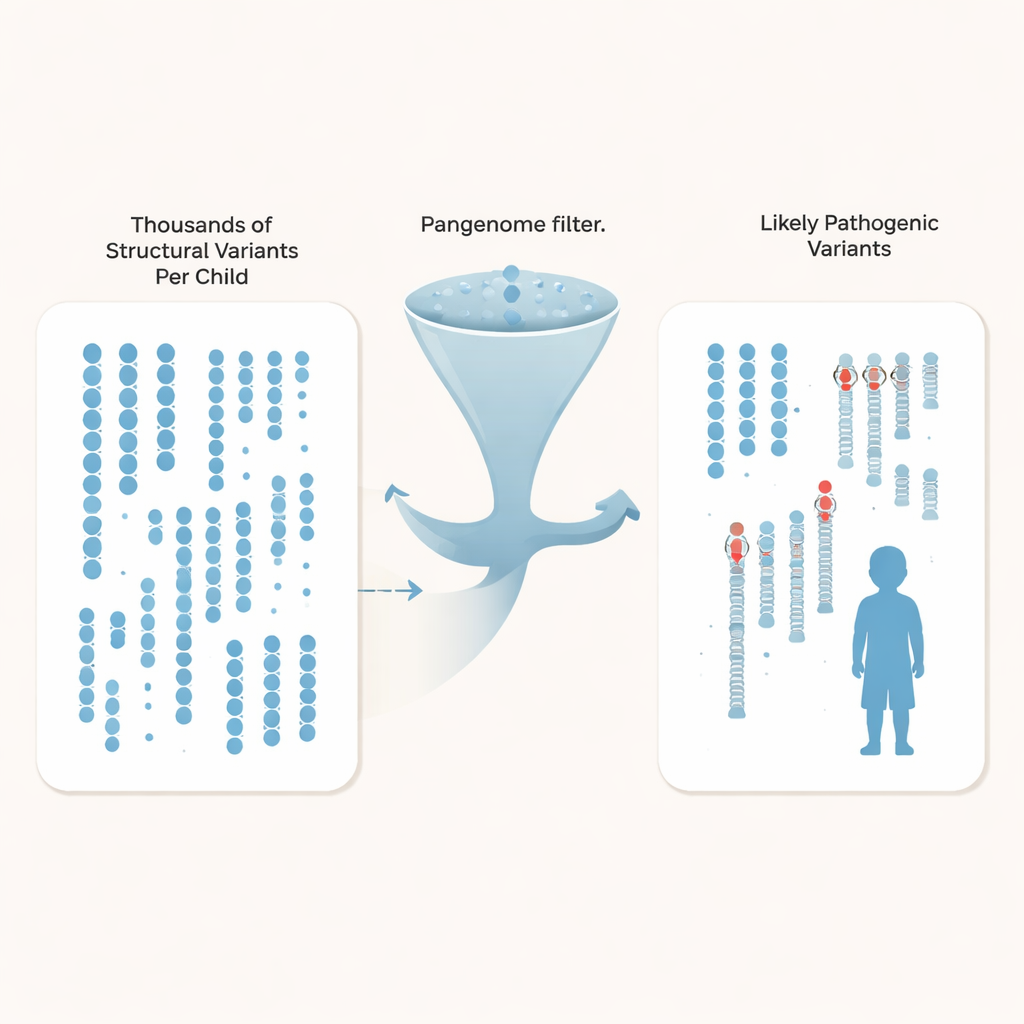
Araştırmacılar sadece tek harflik farklılıkları takip etmek yerine yapısal varyantlara—en az 50 baz uzunluğunda olan ve genleri veya bunların kontrol anahtarlarını bozabilen ekleme, silme ve yeniden düzenlenmelere—odaklandı. Her çocukta ortalama 27.000 civarında böyle varyant vardı, ancak bunların büyük çoğunluğu nüfus içinde paylaşılan zararsız arka plan farklılıklarıydı. Otizmli aileleri, çeşitli kökenlerden gelen yüzlerce derinlemesine dizilenmiş pangenom kontrol genomuyla karşılaştırarak ekip, her çocuk için ortak yapısal varyantların %97’sinden fazlasını filtreleyebildi; böylece her genoma yaklaşık 600 nadir aday kaldı ve en büyük kontrol seti kullanıldığında bu sayı yaklaşık 200’e kadar düşebildi.
Bilinen risk genlerinde kaçırılan mutasyonları bulmak
Arama alanı önemli ölçüde daraltıldıktan sonra yazarlar birkaç kanıt hattını birleştirdi: bilinen otizm ve nörogelişimsel bozukluk genleri, gelişmekte olan insan korteksinde aktif düzenleyici bölgeler ve her aile içindeki kalıtım desenleri. Önceki testlerin atladığı üç açıkça patojenik mutasyonu ortaya çıkardılar. Bunlar arasında sinaps işlevi için önemli olan SYNGAP1 geninde yeni bir durdurma kodonu ve hasta birden fazla klinik testten geçmiş olmasına rağmen Rett sendromunun ana geni MECP2’nin son ekzonunu kesen bir silinme yer alıyordu. Ayrıca MECP2 ile etkileşen bir gen olan TBL1XR1’de hastalığa yol açan bir değişikliği doğruladılar. Toplamda, genellikle kalıtsal ve beyinle ilişkili genlerin yakınlarındaki düzenleyici bölgelerde bulunan dokuz ek yapısal varyantı daha güçlü ileri işlevsel testler için aday olarak vurguladılar.
Çalışmanın bulamadıkları—ve bunun neden yine de önemli olduğu
Bu derin aramaya rağmen, yazarlar otizmli çocuklarda etkilenmemiş kardeşlerine kıyasla genel olarak belirgin bir yapısal varyant fazlalığı görmediler; en azından bu sınırlı örneklem büyüklüğünde. Ancak etkilenen kızlarda X kromozomunda daha fazla yapısal değişiklik olduğuna dair bir ipucu vardı ve neredeyse tamamlanmış X ve Y montajları, aşırı dengesiz X kromozomu deaktivasyonu gibi alışılmadık desenleri tespit etmelerine izin verdi. Bu özellikler daha fazla aile incelendikçe önemli ipuçları haline gelebilir. Kritik olarak, çalışma uzun-okuma dizilemenin, özellikle genomun zor kısımlarında ve gen aktivitesini ince ince ayarlayan kontrol bölgelerinde, kısa-okuma yöntemlerinin kaçırdığı patojenik varyantları kurtarabileceğini gösteriyor.
Bu, aileler ve gelecekteki testler için ne anlama geliyor
Aileler için anlık etki ölçü olarak mütevazı ama anlamlı: çözülmesi zor olan bu vakalar arasında yaklaşık %6’sı net bir genetik tanı aldı ve neredeyse her beş vakadan biri araştırılmaya değer güçlü yeni aday varyantlar elde etti. Alan için mesaj daha geniş. Daha çeşitli, tam referans genomlar pangenoma eklendikçe ve uzun-okuma dizileme daha erişilebilir oldukça, klinisyenler ortak yapısal değişiklikleri hızla eler ve her hasta için nadir, potansiyel olarak zararlı bir dizi varyanta odaklanabilir. Bu dönüşüm zamanla bugünün birçok “çözülmemiş” otizm vakasını, altta yatan biyolojinin—ve destek ile tedavi yollarının—çok daha iyi anlaşıldığı vakalara dönüştürebilir.
Atıf: Sui, Y., Lin, J., Noyes, M.D. et al. Using the linear references from the pangenome to discover missing autism variants. Nat Commun 17, 1681 (2026). https://doi.org/10.1038/s41467-026-68378-4
Anahtar kelimeler: otizm genetiği, uzun-okuma dizileme, yapısal varyantlar, insan pangenomu, Rett sendromu