Clear Sky Science · tr
FLT3-ITD akut myeloid lösemi hücre modellerinde kabozantinib direncini hafifletmek için metabolik imzaların modüle edilmesi
Neden kanser tedavisi için önemli
Birçok modern kanser ilacı tümördeki tek bir hatalı proteine odaklanacak şekilde tasarlanır. Bu hedefe yönelik ilaçlar çarpıcı remisyonlar tetikleyebilir, ancak kanserler sıklıkla uyum sağlayıp yeniden büyümenin yollarını bulur. Bu makale, bir tür kan kanseri olan akut myeloid löseminin (AML) bu tür hedefe yönelik ilaçlardan biri olan kabozantinibe karşı nasıl direnç geliştirdiğini ve kanser hücrelerinin enerji kullanımını yeniden düzenlemenin bu direnci alt etmede doktorlara nasıl yardımcı olabileceğini inceliyor.
Hedefe yönelik bir ilacı atlatmayı öğrenen lösemi hücreleri
Araştırmacılar, özellikle agresif seyirle ilişkilendirilen bir büyüme sinyali anahtarı olan FLT3-ITD mutasyonunu taşıyan AML hücrelerine odaklandı. Kabozantinib, zaten bazı solid tümörlerde kullanılan bir oral ilaç, laboratuvarda FLT3 kaynaklı lösemi hücrelerini güçlü şekilde bloke edebiliyor. Hastalarda zaman içinde ne olduğunu modellemek için ekip, iki FLT3-mutant AML hücre hattını kademeli olarak artan kabozantinib dozlarına maruz bıraktı; bazı hücreler hayatta kalıp yeniden büyümeye başlayana dek. Bu yeni hücre popülasyonları, Molm13-XR ve MV4-11-XR olarak adlandırıldı ve orijinal “ebeveyn” hücrelerine göre çok daha yüksek kabozantinib konsantrasyonlarına tolerans gösterebildi. Ayrıca iki diğer onaylı FLT3 hedefli ilaç olan sorafenib ve quizartinibe karşı daha az duyarlı hale gelirken, farklı bir inhibitör olan gilteritinibe karşı kırılganlıklarını korudular.
Kanserin hayatta kalmasına yardımcı genetik ayarlamalar
İnceleme yapıldığında, bu ilaç dirençli lösemi hücrelerinin FLT3 geninde yeni değişiklikler taşıdığı görüldü. Her iki dirençli hattın da FLT3 kinaz domaininin kritik bir bölgesinde, D835Y olarak adlandırılan aynı nokta mutasyonunu edindiği belirlendi; bu bölge birkaç ilaca karşı direnç için bilinen bir sıcak noktadır. MV4-11-XR hattı ayrıca FLT3’ten bir ekzonun tamamını kaldıran alışılmadık 1.3 kilobazlık bir delesyon kazandı; bu, ilacın bağlanması için önemli bir bölümün silinmesine yol açtı. Bu değişiklikler uzun süreli kabozantinib maruziyeti sırasında seçilmiş gibi görünüyordu: FLT3’ün mutant versiyonları dirençli hücrelerde başlangıç popülasyonuna göre çok daha yaygın hale geldi. Aynı zamanda FLT3’ün aşağı akışında yer alan ERK, STAT5 ve AKT gibi ana sinyal yolları daha güçlü şekilde aktifleştirilmişti; bu da dirençli hücrelerde daha hızlı büyüme ve daha fazla koloni oluşumunu destekledi.
Kanser hücreleri yakıt sistemlerini değiştiriyor
Ekip daha sonra direncin yalnızca genetikle mi yoksa hücrelerin enerji kaynaklarıyla mı ilişkili olduğunu sordu. RNA dizileme ve özel metabolik testler kullanarak tutarlı bir desen buldular: kabozantinib dirençli hücreler, oksijen bolken bile hücre sıvısında glikozun hızlı parçalanması olan glikolize çok daha fazla dayanıyordu. Bu hücreler daha fazla glukoz aldı, daha fazla laktat üretti, GAPDH adlı anahtar bir enzimin aktivitesi yüksekti ve birden çok glikolizle ilişkili genin ekspresyonunu artırdılar. Buna karşılık, daha verimli enerji üretimini destekleyen mitokondriler daha az aktif ve daha az sayıdaydı. Oksijen kullanımı ölçümleri hem temel hem de en yüksek mitokondriyal solunumun azaldığını ve hücre içi reaktif oksijen türlerinin yükseldiğini gösterdi; bu durum mitokondrilerin stres altında ve düşük performanslı olduğunu işaret etti.
Metabolik anahtarı yeniden çeviren ilaçları bulmak
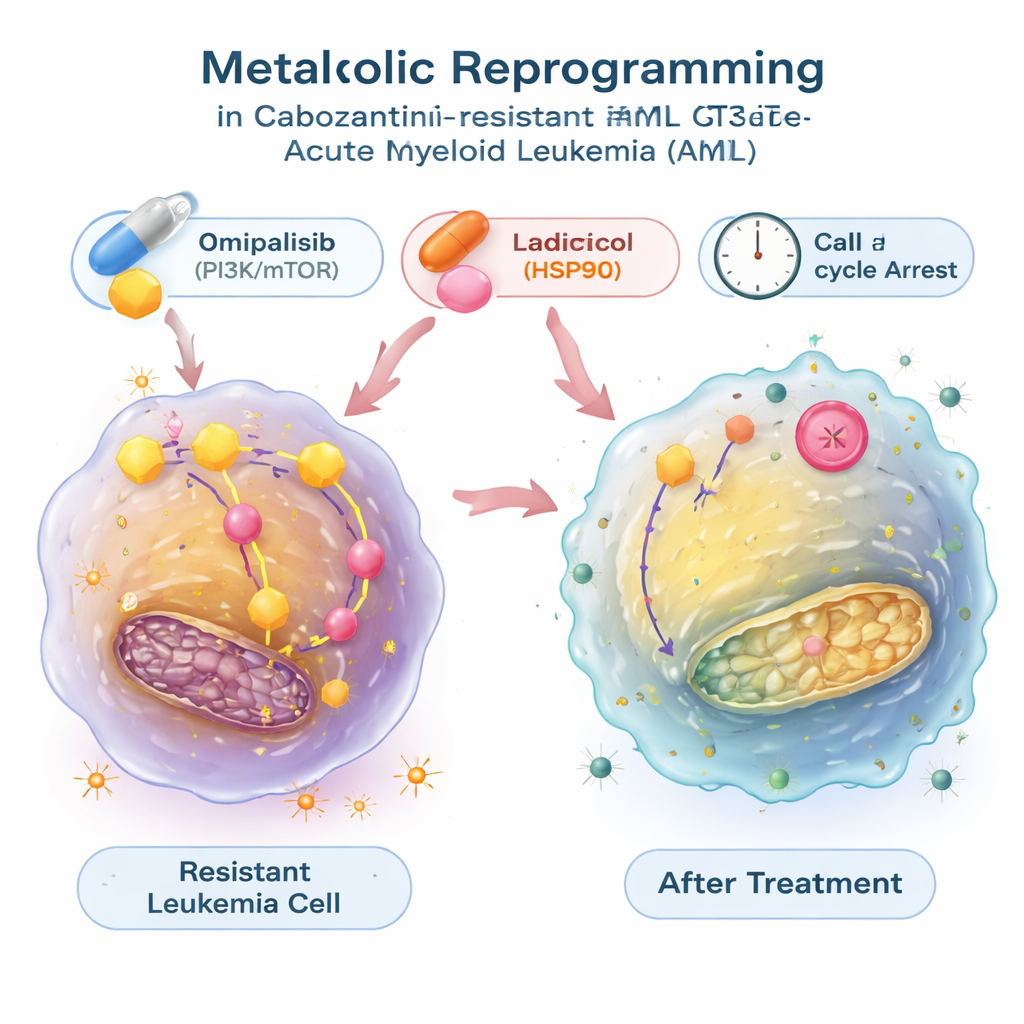
Enerji kaymasının tersine çevrilip çevrilemeyeceğini görmek için araştırmacılar, gen ifadesi paternlerini binlerce bileşiğin etkileriyle ilişkilendiren büyük bir açık veri tabanını kullandı. Dirençli lösemi hücrelerinin metabolik imzasını karşıtlaştırması tahmin edilen ilaçları aradılar ve HSP90 adlı bir protein şaperonunu bloke eden radicicol ile büyüme ve metabolizmayı kontrol eden PI3K/mTOR sinyal yolunu inhibe eden omipalisibi öne çıkardı. Laboratuvar testlerinde her iki molekül de sadece dirençli hücrelerin büyümesini yavaşlatmakla kalmadı, aynı zamanda aşırı aktif glikolizi de azalttı; glukoz alımını ve laktat salınımını normale döndürdü ve glikolizle ilişkili genlerin ifadesini düşürdü. Bu bileşikler lösemi hücrelerini hücre döngüsünün dinlenme fazına itti ve radicicol durumunda aynı zamanda belirgin programlı hücre ölümünü tetikledi. Kabozantinib ile kombinasyon halinde omipalisib — ve bir modelde radicicol — sinerjik etki göstererek ilaç dirençli hücreleri öldürmeyi kolaylaştırdı.
Gelecekteki lösemi tedavileri için anlamı
Uzman olmayanlar için mesaj şudur: lösemi hücreleri hedefe yönelik bir ilaçtan yalnızca doğrudan hedefini mutasyona uğratarak kaçmaz, aynı zamanda enerji üretim ve kullanım biçimlerini değiştirerek da kaçış yolları yaratır. Çalışma, kabozantinib dirençli AML hücrelerinin mitokondrilerini geri planda bırakarak bir “şeker yakma” stratejisi benimsediğini gösteriyor. Omipalisib veya HSP90 inhibitörleri gibi bu yeniden düzenlenmiş metabolizmayı destekleyen yolları hedefleyerek kabozantinibe ve benzeri tedavilere duyarlılığı geri kazandırmak mümkün olabilir. Bu bulgular hücre modellerinden elde edilmiş olsa da, hedefe yönelik kanser ilaçlarını metabolizmayı modüle eden ajanlarla eşleştirmenin FLT3-mutant AML’de direnci geciktirmek veya aşmak için umut verici bir yol olabileceğini düşündürmektedir.
Atıf: Fu, YH., Ng, K.M., Tseng, CY. et al. Modulating metabolic signatures to mitigate cabozantinib resistance in FLT3-ITD acute myeloid leukemia cell models. Cell Death Discov. 12, 98 (2026). https://doi.org/10.1038/s41420-026-02957-8
Anahtar kelimeler: akut myeloid lösemi, ilaç direnci, FLT3 mutasyonu, kanser metabolizması, kabozantinib