Clear Sky Science · sv
Interaktionsbegränsad 3D-molekylgenerering med en diffusionsmodell möjliggör strukturbaserad farmakoformodellering för läkemedelsdesign
Varför det är så svårt att designa bättre läkemedel
Modern läkemedelsupptäckt bygger ofta på att övertala en liten molekyl att passa in i ett protein som en nyckel i ett lås. Men nyckeln måste mer än bara passa: den måste bilda rätt uppsättning av små attraktionskrafter — som svaga elektriska dragningar och vattenavvisande ytor — så att läkemedlet sitter fast starkt och specifikt. Det kemiska universumet är astronomiskt stort, långt bortom vad dagens databaser rymmer, så forskare söker smartare sätt att uppfinna nya nycklar från grunden samtidigt som dessa avgörande kontaktsmönster bevaras.
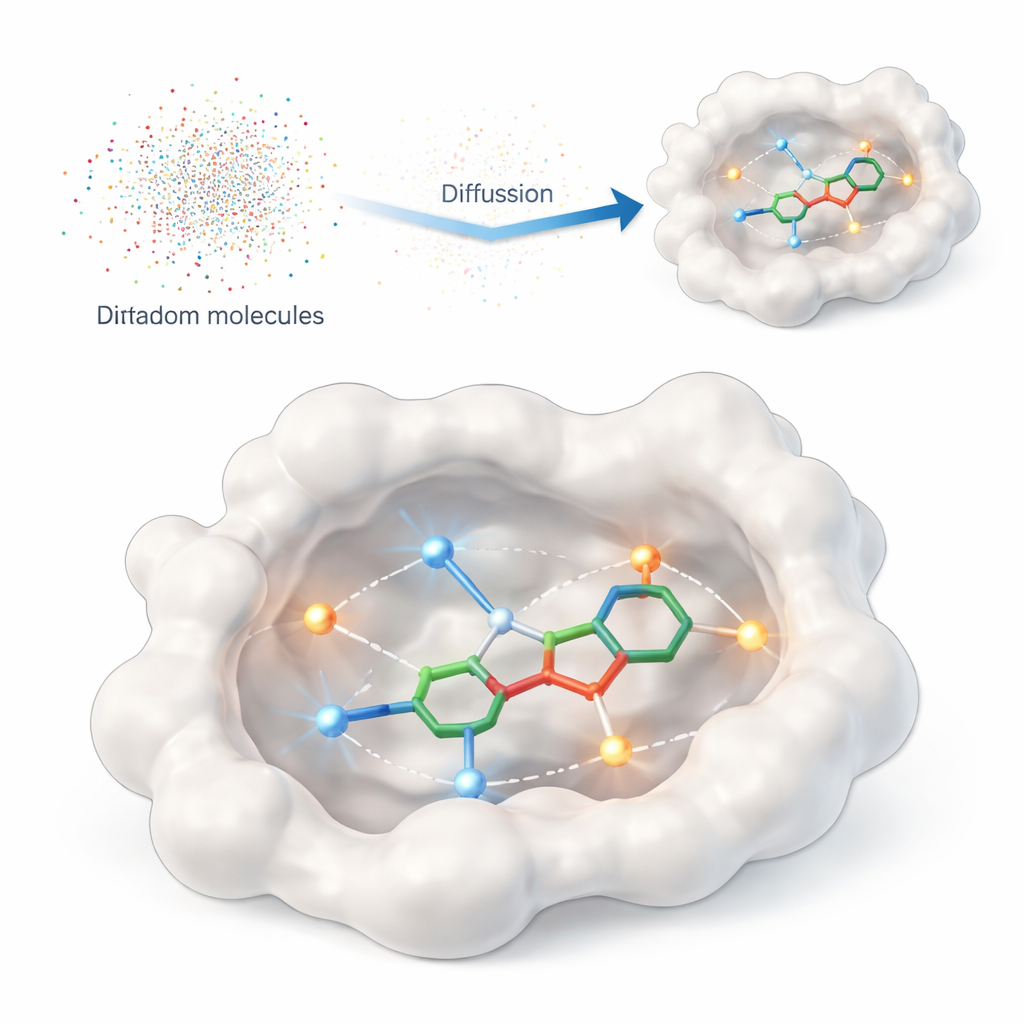
Att lära en dator vad som verkligen betyder något
Denna studie introducerar DiffPharma, ett beräkningsramverk som genererar tredimensionella läkemedelslika molekyler direkt inne i ett proteins bindningsficka. Istället för att be algoritmen söka i stora kataloger av befintliga föreningar skapar DiffPharma nya, atom för atom, styrt av hur de förväntas interagera med proteinet. Metoden bygger på en modern klass av generativa modeller som kallas diffusionsmodeller, vilka börjar från slumpmässigt brus och gradvis ”avbrusar” det till ett strukturerat objekt — i detta fall en 3D-molekyl vilande i proteinets ficka.
Koda proteinets handslag
För att berätta för modellen vad som är viktigt vid proteinsytan representerar författarna nyckelkontakter som små ”interaktionspartiklar” utströdda längs vägarna mellan proteinet och en referensmolekyl. Två vanliga interaktionstyper betonas: vätebindningar, som fungerar som riktade magneter mellan specifika atomer, och hydrofoba kontakter, där oljiga regioner klustrar ihop sig bort från vatten. Separata neurala nätverk lär sig geometrin och kemin för varje interaktionstyp, liksom den övergripande formen av bindningsfickan, och sedan kombinerar en speciell fusionarkitektur dessa synpunkter till en enhetlig, koherent bild som styr molekylgenereringen.
Hur väl efterliknar den verkliga bindningsmönster?
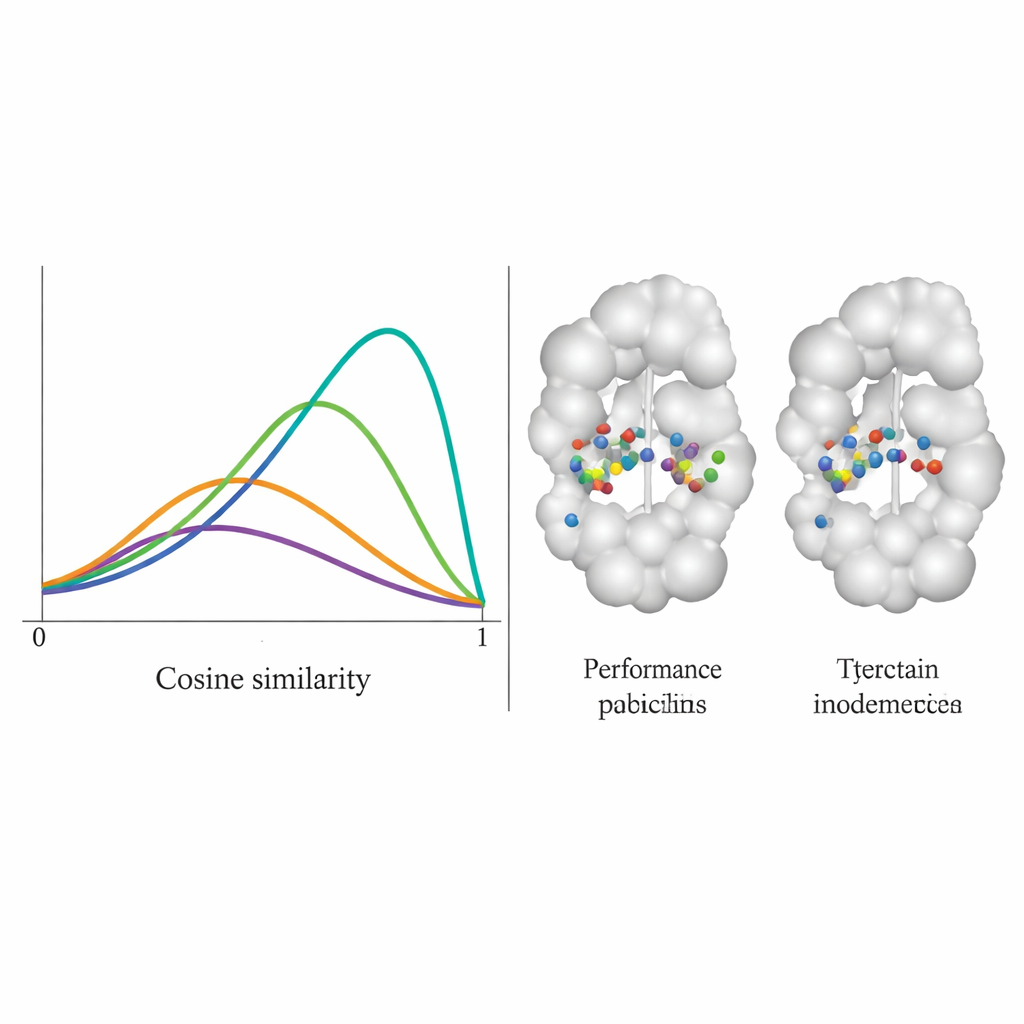
Teamet testade DiffPharma på 100 olika protein–molekylpar och undersökte hur troget nya molekyler återskapade de ursprungliga kontaktsmönstren, rest för rest. De mätte detta med ett cosinuslikhetsscore mellan 0 och 1, där 1 betyder perfekt överensstämmelse. DiffPharmas fördelning toppade runt 0,9, vilket innebär att samma proteinrester i genomsnitt bildade samma typer av nyckelinteraktioner som i referensstrukturerna — avsevärt bättre än sex konkurrerande metoder. Viktigt är att modellen gjorde detta samtidigt som den fortfarande producerade en variation av molekylära former, och de genererade föreningarna bibehöll realistiska bindningslängder, vinklar och övergripande 3D-geometri typisk för verkliga, stabila molekyler.
Från teori till praktiska läkemedelskandidater
Bortom benchmarktester frågade författarna om DiffPharma kunde designa trovärdiga läkemedelskandidater för verkliga mål. För två välstuderade enzymer — AKT-kinas och en β-laktamas kopplad till antibiotikaresistens — genererade metoden molekyler som bevarade de väsentliga interaktionsmönstren hos kända ligander men som ofta använde olika kemiska stommar, en önskvärd form av ”scaffold hopping” inom medicinsk kemi. I en mer krävande fallstudie på SARS‑CoV‑2:s huvudproteas styrdes DiffPharma med specifika interaktionsval och undersöktes sedan med molekylär dynamiksimulering och uppskattningar av bindningsenergi. Molekyler genererade under striktare interaktionsbegränsningar bildade mer stabila komplex och visade ibland mer gynnsamma förutsagda bindningsenergier än en känd referenshämmare. Noterbart återupptäckte systemet till och med den referensföreningen — trots att den aldrig förekommit i träningen — rent utifrån proteinstrukturen och interaktionsinstruktionerna.
Vad detta betyder för framtida läkemedel
För en icke‑specialist kan DiffPharma ses som ett smart, 3D-medvetet ritverktyg för läkemedelsmolekyler: givet formen på en proteinficka och ett önskat mönster av ”handskakningar” föreslår det kemiskt rimliga nycklar som passar och interagerar på rätt sätt. Även om det ännu inte optimerar alla egenskaper ett läkemedel behöver, såsom löslighet eller metabolism, bevarar metoden pålitligt den avgörande kontaktkartan vid proteinsytan och utforskar nya områden av det kemiska rummet bortom nuvarande kataloger. Detta interaktionsstyrda tillvägagångssätt kan hjälpa forskare att snabbare gå från strukturella data om sjukdomsrelaterade proteiner till mångsidiga, realistiska utgångspunkter för experimentell läkemedelsutveckling.
Citering: Sako, M., Yasuo, N. & Sekijima, M. Interaction-constrained 3D molecular generation using a diffusion model enables structure-based pharmacophore modeling for drug design. npj Drug Discov. 3, 8 (2026). https://doi.org/10.1038/s44386-026-00040-x
Nyckelord: strukturbaserad läkemedelsdesign, molekylära generativa modeller, farmakoformodellering, protein–ligandinteraktioner, SARS-CoV-2 huvudproteas