Clear Sky Science · sv
AI-styrd konkurrerande dockning för virtuell screening och prediktion av föreningars effektivitet
Smartare sökningar efter nya läkemedel
Att hitta nya läkemedel är lite som att leta efter en nål i en höstack bestående av miljontals molekyler. Denna studie visar hur senaste framsteg inom artificiell intelligens kan göra den sökningen snabbare och billigare genom att hjälpa forskare förutsäga vilka molekyler som mest sannolikt fäster vid ett sjukdomsrelaterat protein och faktiskt fungerar som läkemedel. Istället för att testa en kemikalie i labbet i taget använder författarna AI-modeller för att låta molekyler tävla virtuellt mot varandra och låta vinnarna stiga till toppen.
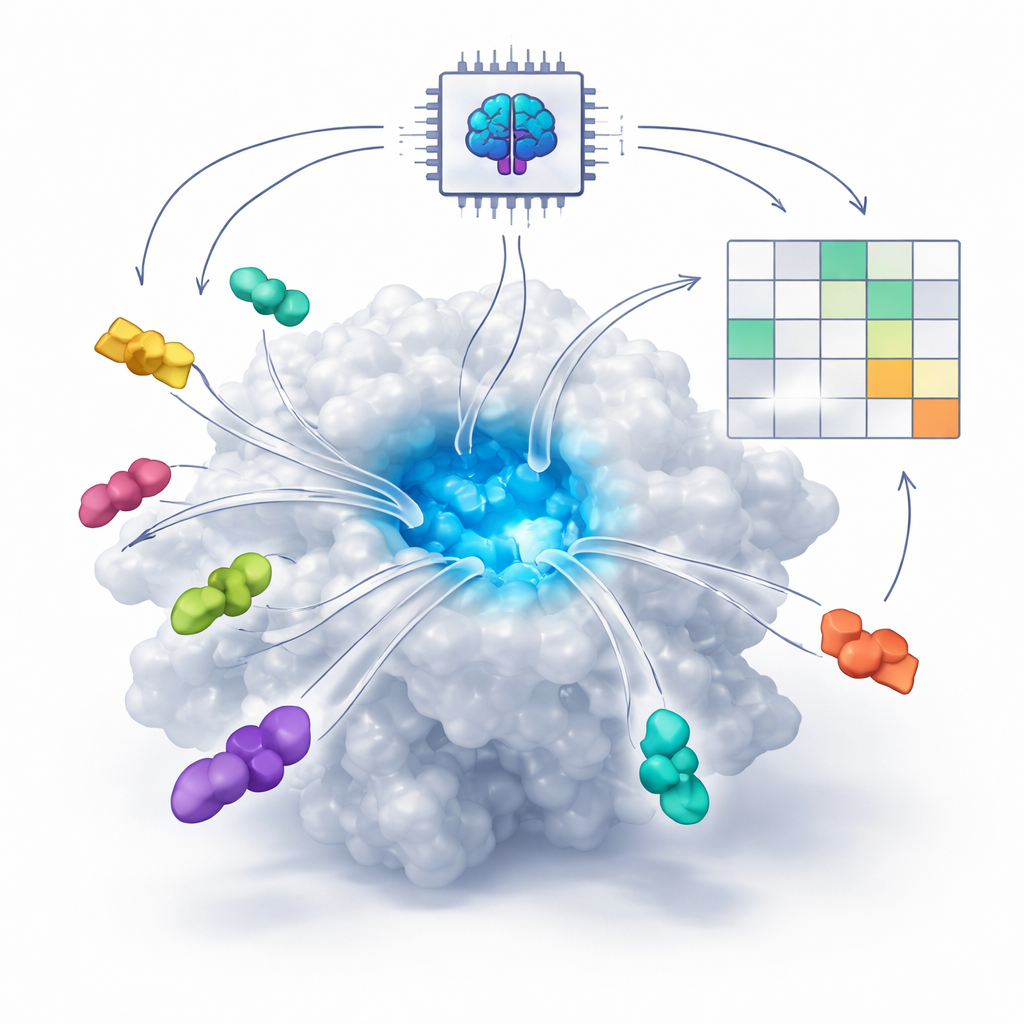
Hur AI lär sig se molekylära lås-och-nyckel-passar
Många moderna läkemedel verkar genom att passa in i små fickor på proteiner, ungefär som en nyckel i ett lås. Traditionellt försökte datorprogram förutsäga denna passform med fysikaliska ekvationer som uppskattar krafter mellan atomer. Under de senaste åren har dock nya AI-system, så kallade diffusionsbaserade co-folding-modeller — såsom AlphaFold3 och Boltz — lärt sig från enorma mängder kända protein–molekylstrukturer. Dessa system kan nu ”föreställa” sig hur ett protein och ett potentiellt läkemedel kan vikas ihop i tre dimensioner, även när ingen experimentell struktur finns. Den centrala frågan författarna tar upp är om dessa AI-verktyg kan göra mer än att bara rita trovärdiga bilder — kan de också skilja bra läkemedel från dåliga?
Riktiga bindare kontra bluffmakare
Teamet testade först 16 välstuderade proteiner plus ett mer komplext bakteriellt enzym kallat DNA-gyras. För varje protein bad de AI-modellerna placera både kända aktiva hämmare och en uppsättning orelaterade ”off-target”-molekyler i samma bindningsficka. Istället för att lita på en enda prediktion undersökte de hur konsekvent AI:n placerade varje molekyl över många körningar. Sanna hämmare tenderade att återvända till samma plats och orientering gång på gång och klustrade inom några biljonredelar av en meter från varandra. Inaktiva molekyler vandrade mer och hamnade ofta längre från fickan. Denna enkla idé — konvergens i position — visade sig vara en stark signal för att en förening verkligen passar sitt proteinmål.
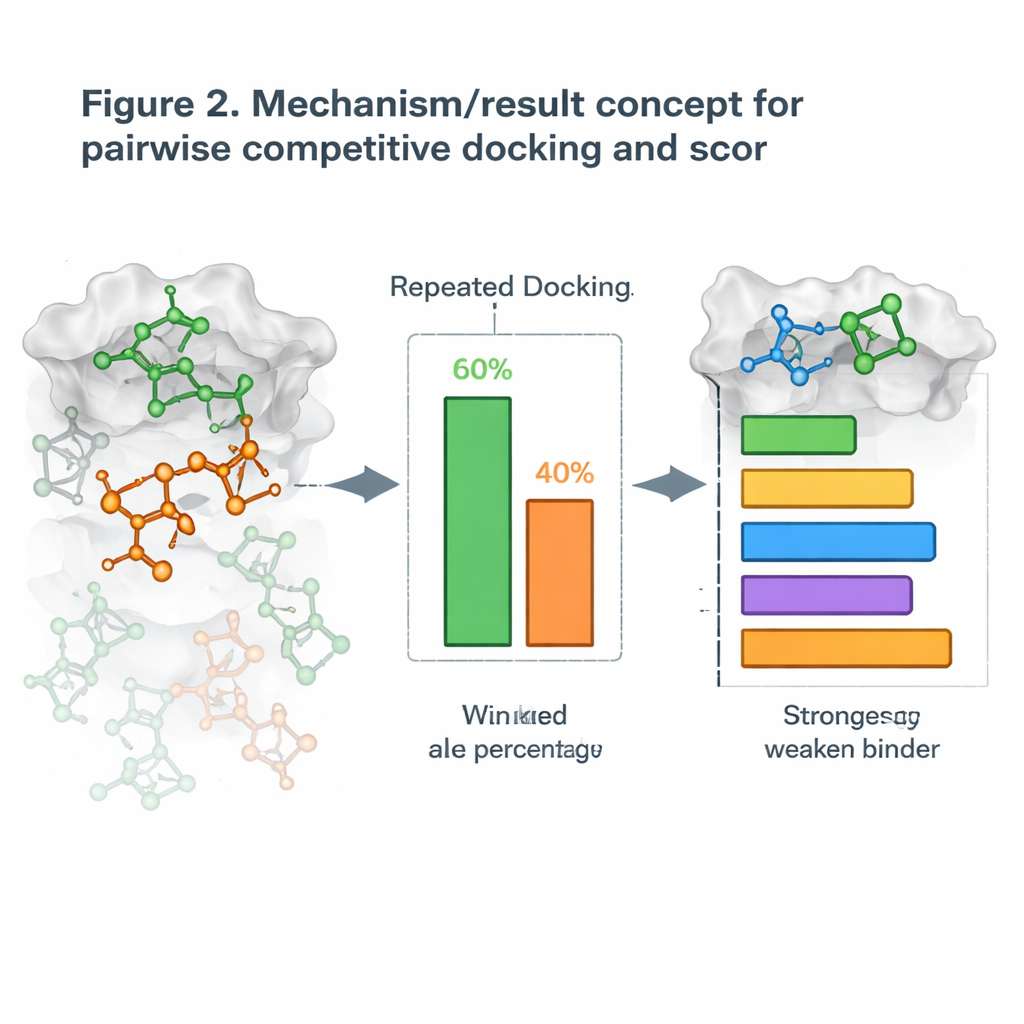
Att göra dockning till en en-mot-en-tävling
Byggt på detta introducerade författarna en ny strategi som de kallar parvis konkurrerande dockning. Istället för att docka en molekyl i taget dockar de två kandidater tillsammans med proteinet och låter dem ”tävla” om samma ficka. Efter många upprepade körningar förklaras den molekyl som oftare upptar platsen som vinnare i det mötet. Genom att köra alla möjliga parningar bygger de en vinst–förlust-tabell och beräknar en Competitive Docking Score för varje molekyl, ungefär som att ranka spelare i en round-robin-turnering. När dessa poäng jämfördes med verkliga mätningar av hur starkt molekylerna blockerar sina mål, stämde rankningarna ofta väl överens, med vissa proteinsystem som visade nästintill perfekt överensstämmelse.
Från virtuella screeningar till att designa bättre antibiotika
DNA-gyras, ett enzym som är nödvändigt för bakterier, fungerade som ett detaljerat testfall. Detta protein har flera läkemedelsfickor som riktas av olika klasser av antibiotika, inklusive de allmänt använda fluoroquinolonerna. AI-modellerna kunde oftast placera varje läkemedelsklass i sin korrekta ficka, och de konkurrerande dockningspoängen följde ungefär deras uppmätta potens. Författarna skalade sedan upp till en virtuell screening av mer än 3 000 godkända läkemedel för att avgöra vilka molekyler som bäst konkurrerade om fluoroquinolon-fickan. Deras tvåstegsstrategi — först använda ”alla-på-en-gång”-konkurrens för att välja troliga vinnare, sedan filtrera efter hur tätt de klustrade i fickan — berikade kraftigt verkliga fluoroquinoloner samtidigt som svagare kandidater avskedades. Slutligen använde de en AI-driven molekylgenerator för att föreslå nya fluoroquinolonliknande strukturer och tillämpade konkurrerande dockning för att hitta ett fåtal med ännu bättre predikterad bindning och acceptabla läkemedelsliknande egenskaper.
Löften, begränsningar och vad det betyder för patienter
Studien visar att moderna AI-modeller kan göra mer än att rita trovärdiga protein–läkemedelsstrukturer: när de körs i ett konkurrensramverk kan de hjälpa till att ranka föreningar på ett sätt som ofta speglar verkliga experimentella data. Detta ersätter inte laboratoriearbete — prestandan beror fortfarande starkt på det specifika proteinet, vissa fickor förutsägs felaktigt och AI-modeller kan misslyckas för mycket stora eller ovanliga molekyler. Men när dessa modeller och deras träningsdata förbättras kan metoder som parvis konkurrerande dockning göra den tidiga läkemedelsupptäckten mycket mer effektiv. För patienter kan det i förlängningen innebära snabbare utveckling av riktade läkemedel, inklusive nya antibiotika som håller jämna steg med resistenta bakterier.
Citering: Mirgaux, M., Barcelli, V., Chua, A.C.Y. et al. AI-guided competitive docking for virtual screening and compound efficacy prediction. npj Drug Discov. 3, 6 (2026). https://doi.org/10.1038/s44386-026-00039-4
Nyckelord: AI-läkemedelsupptäckt, virtuell screening, molekylär dockning, protein-ligandbindning, antibiotikadesign