Clear Sky Science · sv
Energieffektiv vetenskaplig beräkning med kemiska reservoarer
Varför det spelar roll att göra kemi till beräkning
Moderna superdatorer förbrukar enorma mängder elektricitet för att simulera klimat, utforma nya läkemedel eller träna artificiell intelligens. När vi närmar oss de fysiska gränserna för traditionella kretsar blir det svårare och dyrare att pressa fram mer prestanda per watt. Denna artikel utforskar en radikalt annorlunda väg: att använda verkliga kemiska reaktioner som motorn i vetenskaplig beräkning. Genom att betrakta molekyler och deras interaktioner som datorns rörliga delar skissar författarna på hur framtida maskiner skulle kunna lösa komplexa ekvationer med avsevärt mindre energi än dagens digitala hårdvara.
Från levande celler till kemiska räknemaskiner
Levande celler är mästare på problemlösning. De jonglerar ständigt med tusentals reaktioner för att anpassa sig, växa och överleva, samtidigt som de använder förvånansvärt lite energi. I centrum för detta beteende finns nätverk av kemiska reaktioner—sammanlänkade reaktioner vars hastigheter och koncentrationer ändras över tid. Dessa nätverk kan beskrivas med ordinära differentialekvationer, samma matematiska språk som används för att modellera allt från epidemier till turbulenta flöden. Insikten bakom detta arbete är att om kemin redan följer dessa ekvationer, kan vi kanske utnyttja den direkt för att utföra de beräkningar som forskare i dag gör på kiseldatorer.
Hur ekvationer blir reaktionsnätverk
Författarna presenterar ChemComp, ett programvaruramverk som tar ett system av differentialekvationer och systematiskt omvandlar det till ett abstrakt nätverk av reaktioner. ChemComp använder modern kompilatorteknik för att bryta ner ett matematiskt problem i mönster som kan representeras av idealiserade reaktioner, och organiserar dem sedan till ett nätverk med väl definierade arter, kopplingar och hastigheter. Dessa abstrakta reaktioner motsvarar ännu inte verkliga molekyler, men utgör en ritning för en kemisk dator. Ramverket kan sedan söka i biokemiska reaktionsdatabaser för att hitta verkliga reaktionsmotiv som beter sig likartat, och föredrar alternativ som är praktiska, säkra och potentiellt energieffektiva i laboratoriemiljö.
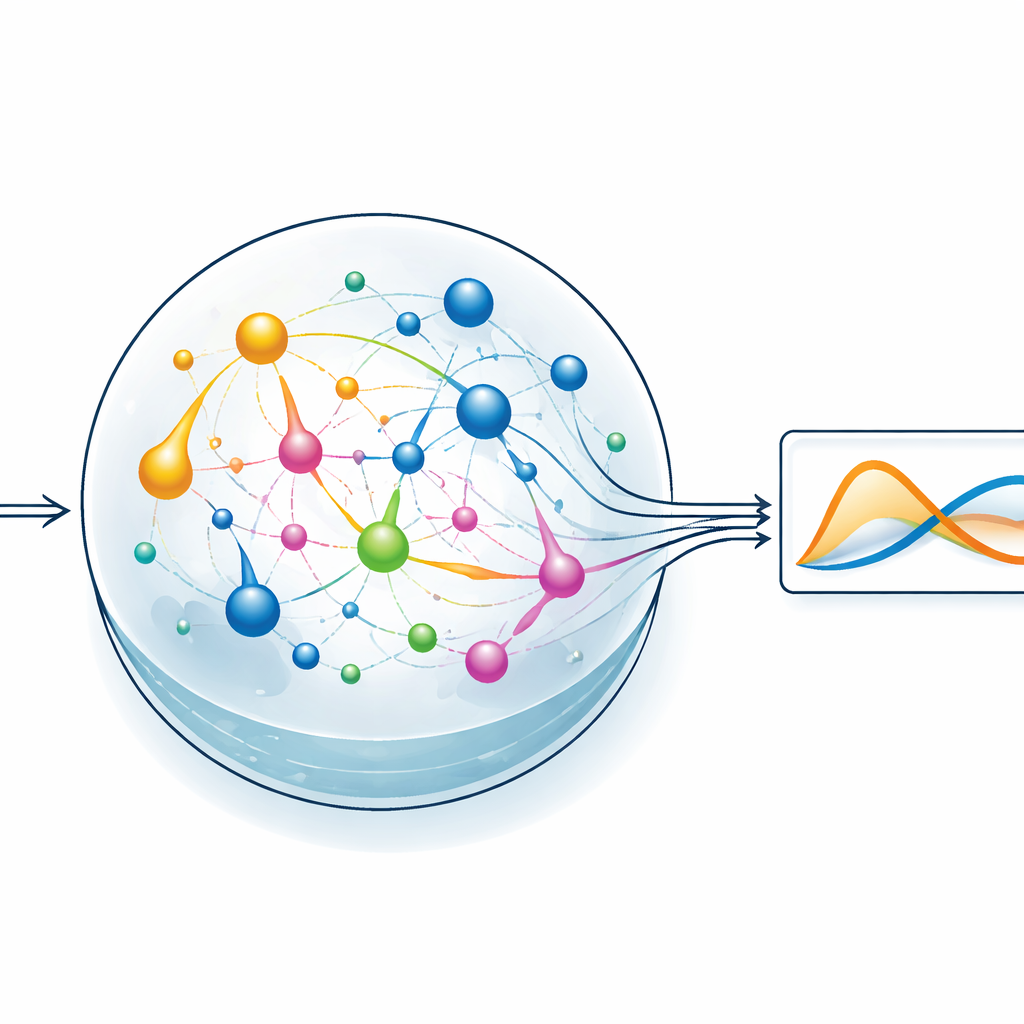
Låta ett kemiskt reservoar göra det tunga arbetet
För att testa idén fokuserar teamet på en typ av maskininlärning kallad reservoarberäkning. Här omvandlar ett fast, dynamiskt system en insignalsignal till ett rikt, indraget mönster av intern aktivitet, och endast ett enkelt avläsningslager tränas för att producera önskat utfall. I ChemComp-versionen är reservoaren en uppsättning reaktioner i ett välomrört kärl; de förändrade koncentrationerna av kemikalier utgör de interna tillstånden. Författarna kompilerar ett klassiskt tvåvariabelsystem känt som Sel’kov–Schnakenberg-modellen—ursprungligen använt för att studera oscillationer i metabolismen—till kandidatreaktionsnätverk. De simulerar sedan hur dessa nätverk svarar över tid när de drivs av flöden av kemikalier in och ut ur kärlet, och använder enkel linjär regression för att kombinera koncentrationsspåren till en approximation av mållösningen.
Test av enkla och mer komplexa kemiska nätverk
Forskarna jämför två kandidatreservoarer: en med bara två kemiska arter och två reaktioner, och en annan med fem arter och fem reaktioner. Båda nätverken förses med lämpliga startkoncentrationer och flödeshastigheter och simuleras medan de körs. Även det mindre systemet kan ungefär återskapa det oscillerande beteendet i målekvationerna, men det större nätverket presterar märkbart bättre och minskar felet både under träning och testning. Genom att skanna över olika initiala koncentrationer och reaktionshastighetskonstanter kartlägger författarna områden där det kemiska systemet mest liknar den önskade dynamiken. Varje reaktion fungerar i praktiken som en basfunktion i ett kurvanpassningsproblem: ju fler varierade reaktioner som finns tillgängliga, desto lättare blir det att approximera komplexa beteenden, på bekostnad av ökad systemkomplexitet.
Vägen mot lågenergiberäkning i laboratoriet
Utöver simuleringar blickar artikeln framåt mot praktiska enheter. Den diskuterar hur val av reaktion måste balansera energianvändning, möjligheten att styra med enzymer eller katalysatorer, och förmågan att mäta nyckelarter i realtid, till exempel med optiska eller elektro-kemiska metoder. Författarna föreslår att framtida mikrofluidiska plattformar skulle kunna hysa noggrant utvalda reaktionsnätverk, med rumslig kontroll av insignal och inbyggd sensorteknik. Även om många konstruktionsutmaningar återstår—from att kartlägga ekvationer till verklig kemi till att hantera brus och mätbegränsningar—visar studien att måttliga reaktionssystem redan kan efterlikna lösningar av koppade differentialekvationer. För en icke-expert är kärnbudskapet att kemin själv kan fungera som en analog dator och öppna en väg till vetenskapliga beräkningar som bygger på de energieffektiva processer naturen finjusterat under miljarder år.
Citering: Johnson, C.G.M., Bohm Agostini, N., Cannon, W.R. et al. Energy-efficient scientific computing using chemical reservoirs. npj Unconv. Comput. 3, 17 (2026). https://doi.org/10.1038/s44335-026-00053-9
Nyckelord: kemisk beräkning, energieffektiv beräkning, reservoarberäkning, nätverk av kemiska reaktioner, ordinära differentialekvationer