Clear Sky Science · sv
En helhetsram för reaktivitet i heterogen katalys
Varför snabbare katalysatordesign spelar roll
Det moderna samhället förlitar sig på katalysatorer för att framställa bränslen, plaster, gödningsmedel och otaliga vardagsprodukter. Att hitta bättre katalysatorer är dock ofta som att leta efter en nål i en höstack, eftersom varje material kan främja tusentals möjliga mikroskopiska reaktioner samtidigt. Denna artikel presenterar CARE, ett nytt beräkningsramverk som använder smarta regler och maskininlärning för att kartlägga och simulera dessa intrasslade reaktionsnätverk mycket snabbare och mer fullständigt än tidigare. Genom det lovar det att styra renare energitekniker och mer effektiva kemiska processer samtidigt som beräkningskostnaderna minskas kraftigt.
Att reda ut trånga reaktionsvägar
På ytan av en fast katalysator följer inkommande molekyler inte bara en enda prydlig väg från reaktant till produkt. I stället rör de sig genom en labyrint av kortlivade mellanprodukter och konkurrerande vägar. Traditionella datorbaserade metoder förlitar sig på mänsklig intuition för att välja en begränsad uppsättning möjliga steg och använder därefter kvantberäkningar för att utvärdera deras energier. Det fungerar för små nätverk men kollapsar snabbt när systemen blir mer komplexa, och förbiser sällsynta vägar som kan styra långsiktig aktivitet, deaktivering eller selektivitet. CARE angriper denna utmaning genom att automatiskt konstruera mycket stora reaktionsnätverk från enkla byggregler, och säkerställer att alla rimliga bindningsbrytande och bindningsbildande händelser mellan kol, väte och syre inkluderas, även sådana som kemister normalt kan avfärda.
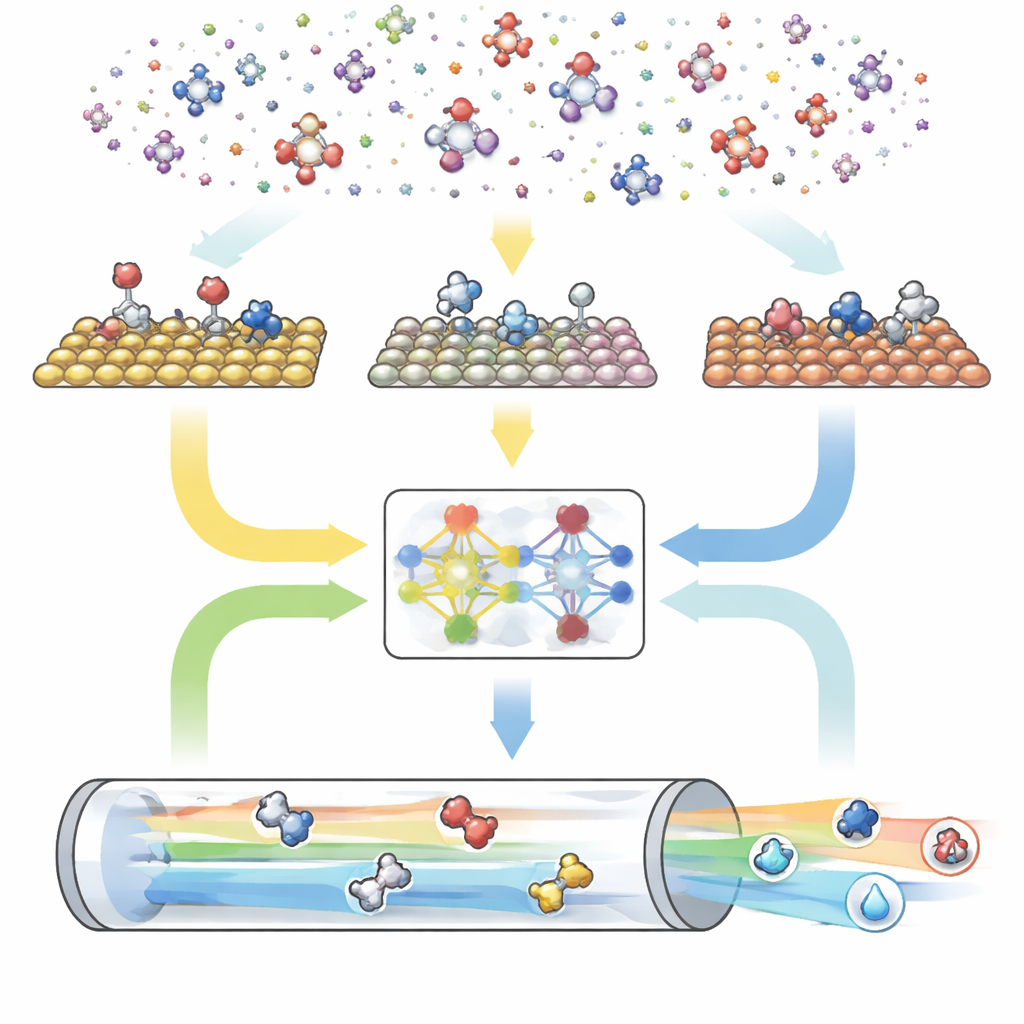
En tredelad digital motor för reaktioner
CARE är uppbyggt som en helhetspipeline med tre huvudmoduler. Först definierar en regelbaserad generator ”kemiskt utrymme” genom att välja maximal antal kol- och syreatomer och sedan tillämpa enkla mallar för att skapa alla matchande molekyler och deras ytbundna former. För det andra använder en energiutvärderingsmodul moderna maskininlärningsmodeller—särskilt ett grafneuronätverk kallat GAME-Net-UQ—för att uppskatta energierna för mellanprodukter och övergångstillstånd på många metallytor. Denna modell behandlar varje struktur som ett nätverk av atomer och bindningar, returnerar både en energi och en osäkerhet, och är korrekt inom några tiondels elektronvolt samtidigt som den förblir lättviktig och snabb. För det tredje använder en mikrokinetisk solver dessa energier för att beräkna hur alla reaktioner fortskrider tillsammans under realistiska förhållanden av temperatur, tryck, spänning och pH, och förutsäger övergripande reaktionshastigheter, ytöverdragningar och produktselektivitet.
Test i verkliga tillämpningar: bränslemolekyler och klimatkemi
För att visa att CARE inte bara är en teoretisk övning tillämpar författarna det på tre industriellt relevanta problem med ökande svårighetsgrad. För metanoldekomposition—en reaktion viktig för vätelagring—genererar de ett måttligt nätverk och utvärderar det över många metaller och kristallplan. CARE bygger upp den välbekanta ”volcanotrenden” i aktivitet och identifierar korrekt ruthenium som en av de bästa aktörerna, i linje med experiment, men till en bråkdel av beräkningstiden som krävs för fulla kvantberäkningar. Därefter går de över till elektrochemisk omvandling av koldioxid på koppar, med fokus på hur trekolprodukter som 1-propanol och propylene uppstår. Genom att inkludera speciella steg som tar hänsyn till protoner, elektroner och lösningsförhållanden fångar CARE hur pH och applicerad spänning förskjuter vägarna och förutsäger korrekt att 1-propanol gynnas framför propylene, vilket speglar tidigare detaljerade studier.
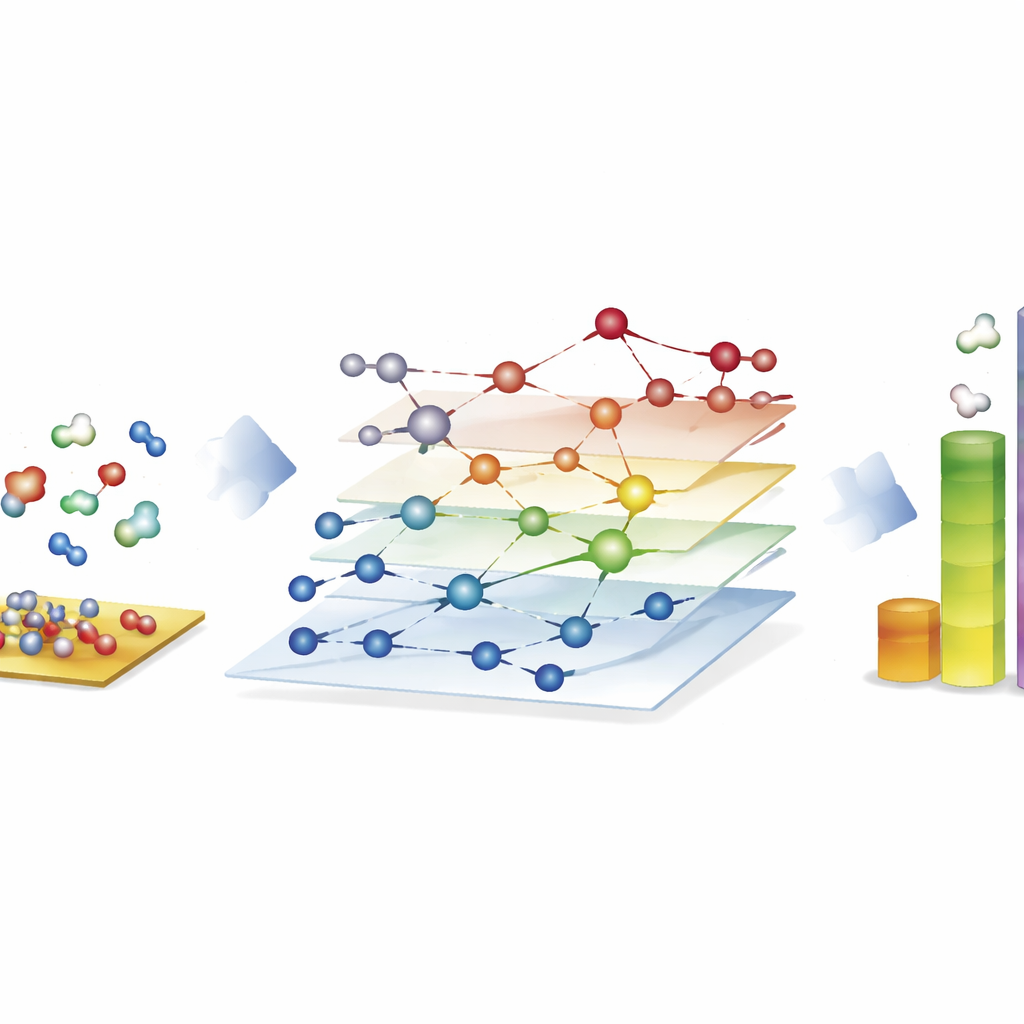
Utforska enorma reaktionsnät för syntetiska bränslen
Den mest slående demonstrationen kommer från Fischer–Tropsch-processen, som omvandlar blandningar av kolmonoxid och väte till långkedjiga kolväten för bränslen och kemikalier. Här konstruerar författarna nätverk med nästan 40 000 ytspecies och cirka 370 000 elementära reaktioner—långt bortom vad traditionella kvantbaserade studier kan utforska fullt ut. Med CARE utvärderar de alla mellanprodukter och nyckelbarriärer på kobolt-, järn-, nickel- och rutheniumytor på bara några timmar på standardhårdvara, en snabbhetsökning på ungefär en miljon gånger jämfört med direkta kvantberäkningar. Mikrokinetiska simuleringar på dessa nätverk reproducerar kända trender: kobolt och järn bildar företrädesvis längre kolkedjor, järn ger mer koldioxid genom sidoreaktioner, och nickel tenderar mot starkare hydrering. Även om vissa detaljer, såsom metanutbyten, förblir ofullständiga, visar ramverket vilka bindningsbildande steg som dominerar kedjetillväxten och belyser var modeller fortfarande behöver förfinas.
Vad detta innebär för framtida katalysatorer
För icke-specialister är huvudbudskapet att CARE erbjuder ett praktiskt sätt att utforska enorma reaktionsutrymmen på katalytiska ytor som tidigare var utom räckhåll. Genom att automatisera nätverksgenerering, koppla in snabba maskininlärnings-”surrogat”modeller för kvanttillståndens energier och lösa den resulterande kinetiken effektivt kan det rangordna kandidatkatalysatorer, identifiera lovande driftförhållanden och upptäcka oväntade vägar med mycket mindre mänsklig bias och beräkningskostnad. Medan författarna noterar återstående utmaningar—såsom bättre hantering av trånga ytor, lösningsmedelseffekter och ännu större nätverk—pekar arbetet mot en framtid där datorer snabbt kan skanna komplexa reaktioner, från koldioxidreduktion till plaståtervinning och uppgradering av biomassa, och vägleda experiment mot de mest lovande idéerna i stället för att lämna upptäckten åt trial-and-error.
Citering: Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng 3, 169–180 (2026). https://doi.org/10.1038/s44286-026-00361-8
Nyckelord: heterogen katalys, reaktionsnätverk, maskininlärning, mikrokinetisk modellering, Fischer–Tropsch-syntes