Clear Sky Science · sv
Integrativ genomisk och litteraturbaserad bedömning av desmoglein 2‑relaterad arytmogen kardiomyopati med validering i en italiensk kohort
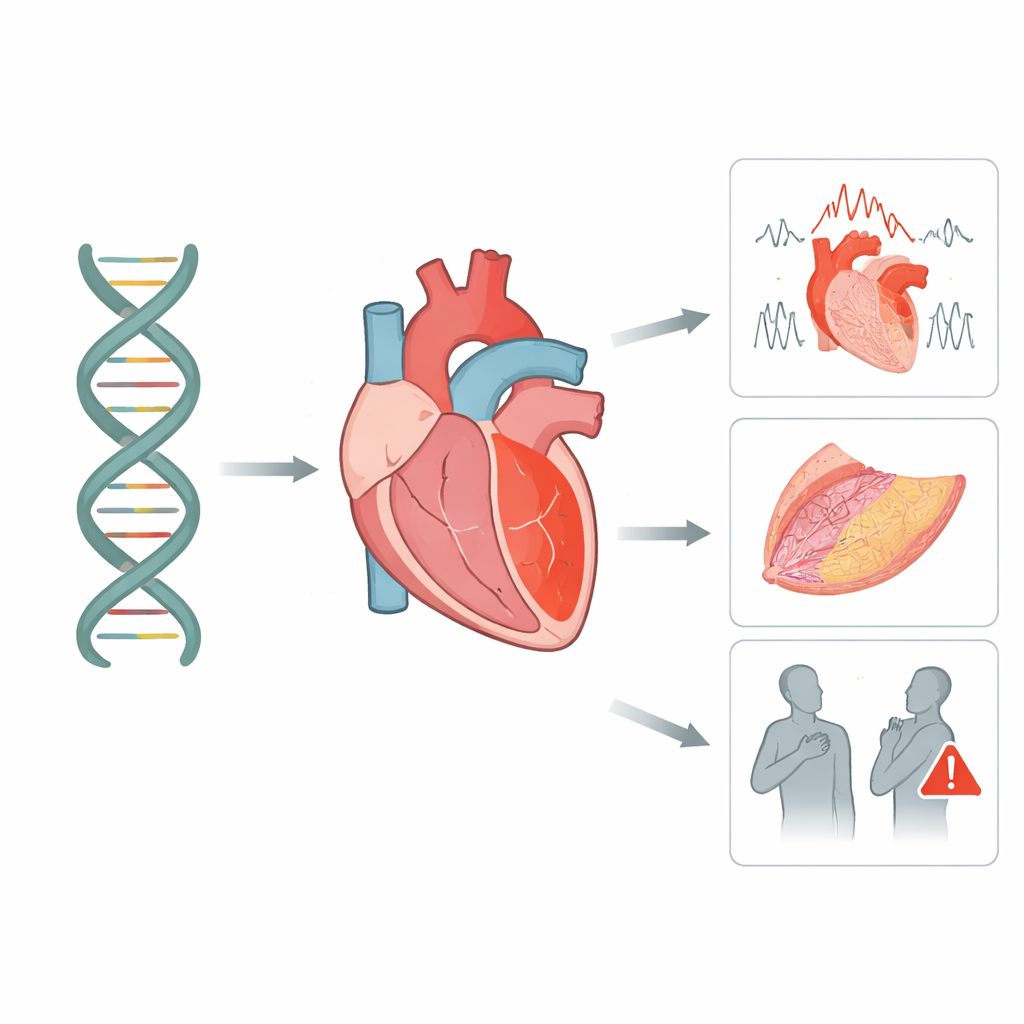
Varför den här hjärtgenen betyder något för familjer
Många akuta hjärtproblem hos unga och i övrigt friska personer är inte slumpmässiga — de är skrivna, åtminstone delvis, i deras DNA. Denna artikel undersöker ett viktigt hjärt‑”lim” i form av proteinet desmoglein‑2 och visar hur små förändringar i dess gen kan försvaga hjärtmuskeln, rubba hjärtats elektriska rytm och öka risken för farliga händelser. Genom att kombinera stora genetiska databaser med en noggrant följd italiensk patientgrupp ger forskarna klarare svar för familjer som undrar vad ett testresultat för denna gen verkligen innebär.
Hjärtats mekaniska lim
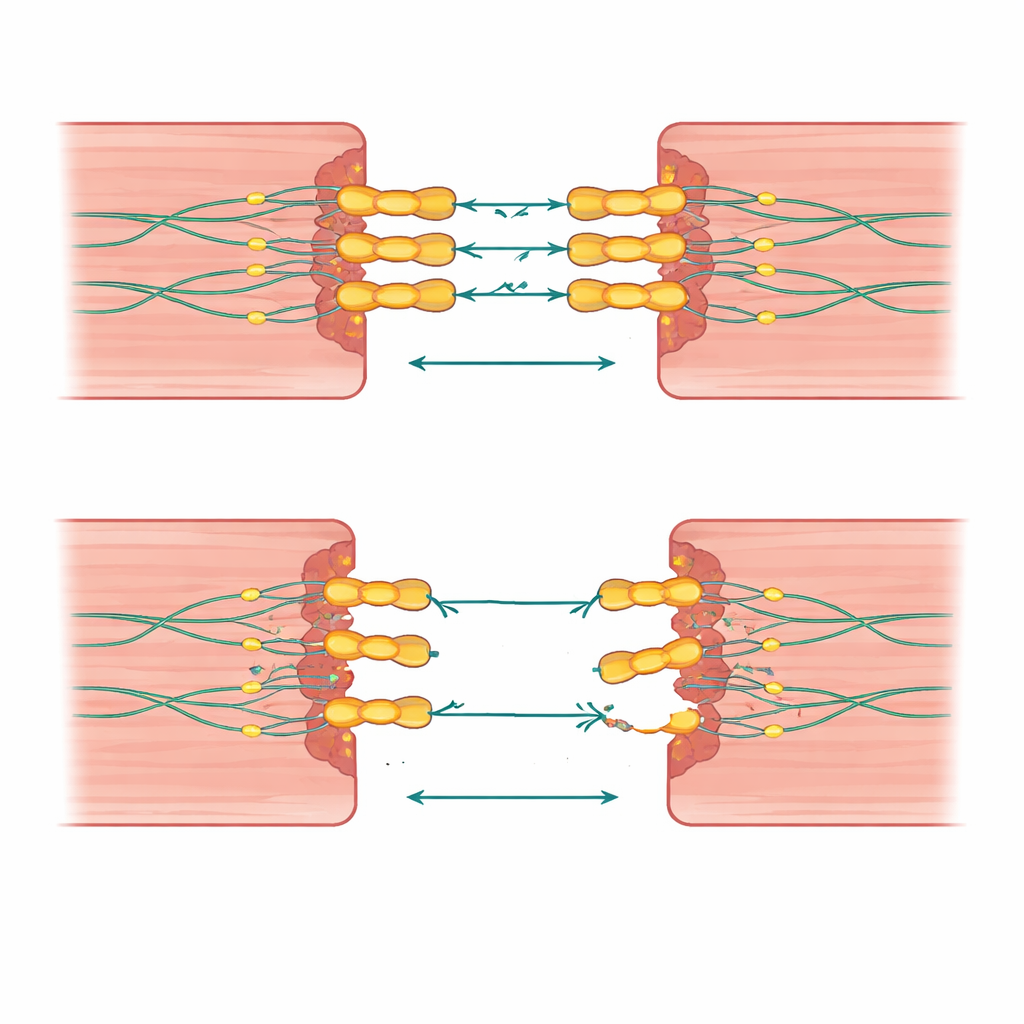
Hjärtmuskelceller måste hålla hårt ihop medan de slår miljontals gånger under en livstid. Desmoglein‑2 är en del av en mikroskopisk nitliknande struktur som låser ihop intilliggande celler så att de kan dra ihop som ett team. Författarna förklarar hur detta protein sträcker sig från cellens utsida, där det griper ett matchande partnerprotein på nästa cell, till insidan, där det hakar i ett stödjande skelett. Eftersom desmoglein‑2 är den enda medlemmen i sin familj som finns i hjärtceller kan allvarlig skada på det inte kompenseras av en reserv, vilket gör hjärtat särskilt sårbart.
Att skilja meningsfulla genförändringar från bakgrundsbrus
Modern sekvensering hittar tusentals skillnader i desmoglein‑2‑genen i populationen, men de flesta orsakar inte sjukdom. Teamet granskade systematiskt 115 publicerade studier och använde två stora offentliga databaser som tillsammans listade mer än 5 000 varianter. Med vedertagna regler inom medicinsk genetik omklassificerade de varje förändring efter hur sannolikt skadlig den är. De fann att verkligt skadliga varianter klustrar i specifika regioner av proteinet — särskilt de yttre segmenten som behöver kalcium för att bilda en rigid bro mellan celler, en kort sträcka som måste klippas för att proteinet ska mogna, och den inre regionen som fäster vid ett annat nyckelprotein i hjärtat. Många andra förändringar förblev ”osäkra”, men en del visade starka indikationer på betydelse och flaggades för närmare uppföljning.
Vad den italienska patientgruppen avslöjar
För att se hur dessa genetiska mönster yttrar sig i verkliga människor studerade forskarna 95 individer i Italien som bar desmoglein‑2‑varianter och undersöktes grundligt med avbildning, rytmtester och långtidsuppföljning. Ungefär hälften uppfyllde strikta kriterier för arytmogen kardiomyopati, ett tillstånd där delar av hjärtmuskeln gradvis ersätts av ärr och fett, vilket skapar förutsättningar för farliga rytmrubbningar. Bland släktingar som bar en variant visade endast cirka fyra av tio faktiskt tecken på sjukdom, vilket understryker att ett positivt gentest inte garanterar sjukdom men signalerar behovet av noggrann övervakning. De med tydlig sjukdom hade en märkbar börda av allvarliga rytmhändelser, medan transplantationer och dödsfall var mindre frekventa men fortfarande förekom.
När en träff inte är nog
En slående insikt från detta arbete är att antalet och kombinationen av genförändringar spelar roll. Personer som ärvt två defekta kopior av desmoglein‑2, eller en desmoglein‑2‑variant plus en förändring i ett besläktat hjärt‑”lim”‑gen, tenderade att bli sjuka tidigare och uppvisa mer utbredd skada på båda sidor av hjärtat. Vissa familjer bar stora deletioner eller duplikationer som tog bort eller fördubblade inte bara desmoglein‑2 utan också närliggande gener, vilket åter kopplade dessa förändringar till aggressiv sjukdom och kluster av plötsliga dödsfall. I kontrast hade många släktingar med endast en enstaka förändring milda eller inga symtom, vilket tyder på att bakgrundsgener och livsstilsfaktorer som fysisk aktivitet kan fälla avgörandet mellan tyst risk och tydlig sjukdom.
Från proteinform till patientrisk
För att koppla DNA‑koden till fysiska effekter använde teamet avancerade 3D‑proteinmodeller för att se hur specifika substitutioner kan lossa desmoglein‑2‑stommen. Förändringar som snedvrider kalcium‑bindande slingor eller bryter viktiga fästpunkter förutspåddes destabilisera proteinet och försvaga cell‑till‑cell‑adhesionen. Dessa strukturella ledtrådar införlivades i klassificeringssystemet och hjälpte till att skjuta vissa gränsfall mot att bedömas som mer sannolikt skadliga eller mer sannolikt ofarliga. Denna bro mellan molekylär modellering och kliniska data förflyttar genetisk testning bortom enkel kodläsning mot en mer funktionell förståelse.
Vad detta innebär för patienter och familjer
För familjer som berörs av arytmogen kardiomyopati erbjuder denna studie både varning och vägledning. Den visar att inte varje desmoglein‑2‑variant är en dom över svår hjärtsjukdom, men att vissa mönster — särskilt multipla träffar eller förändringar i kritiska regioner av proteinet — är kopplade till tidigare och allvarligare problem. Författarna argumenterar för att personer som bär dessa varianter inte bör avfärdas som ”friska tills motsatsen bevisas”, utan istället följas under hela livet med skräddarsydda rytmkontroller och bilddiagnostik. Deras integrativa angreppssätt — som förenar storskalig genetik, detaljerade familjestudier och proteinstruktur — pekar mot mer precisa riskuppskattningar och säkrare rådgivning när en desmoglein‑2‑förändring dyker upp i ett gentest.
Citering: Pinci, S., Celeghin, R., Martini, M. et al. Integrative genomic and literature assessment of desmoglein 2-related arrhythmogenic cardiomyopathy with Italian cohort validation. Commun Med 6, 145 (2026). https://doi.org/10.1038/s43856-026-01416-w
Nyckelord: arytmogen kardiomyopati, desmoglein‑2, ärftlig hjärtsjukdom, genetisk risk, plötslig hjärtdöd