Clear Sky Science · sv
Villkorsstyrd diffusion med lokalitetsmedveten modal anpassning för att generera mångfaldiga proteinkonformationsensembler
Varför proteinrörelse spelar roll
Proteiner i våra celler är inte styva skulpturer; de fungerar mer som små, flexibla maskiner som ständigt förändrar sina former. Dessa formförändringar kan styra hur enzymer katalyserar reaktioner, hur receptorer reagerar på läkemedel och hur signaler flödar genom celler. Ändå visar de flesta välkända bilder av proteiner bara ett ”ögonblicksbild”-struktur, vilket missar det rika ensemble av former som faktiskt finns. Denna artikel presenterar Mac-Diff, en artificiell intelligensmetod som snabbt kan generera många realistiska former för ett givet protein, och hjälper forskare att se inte bara hur ett protein ser ut utan också hur det andas och rör sig.
Från enstaka ögonblicksbilder till rörliga ensembler
I årtionden har forskare förlitat sig på mödosamma experiment eller långkörande molekylär dynamik-simuleringar för att utforska proteinrörelse, vilket båda kan vara långsamma och kostsamma. Genombrottsverktyg som AlphaFold2 förutsäger nu ett proteins mest sannolika 3D-struktur direkt från dess aminosyrasekvens, men återger vanligtvis bara en eller några få föredragna former. Många proteiner, särskilt de som ingår i signalering och allosterisk reglering, intar naturligt flera, löst definierade tillstånd. Författarna menar att för att förstå hur sådana proteiner verkligen fungerar — och för att designa läkemedel som binder till mindre uppenbara, övergående former — behöver vi ett sätt att generera hela ensembler av rimliga konformationer, inte bara en bästa gissning.
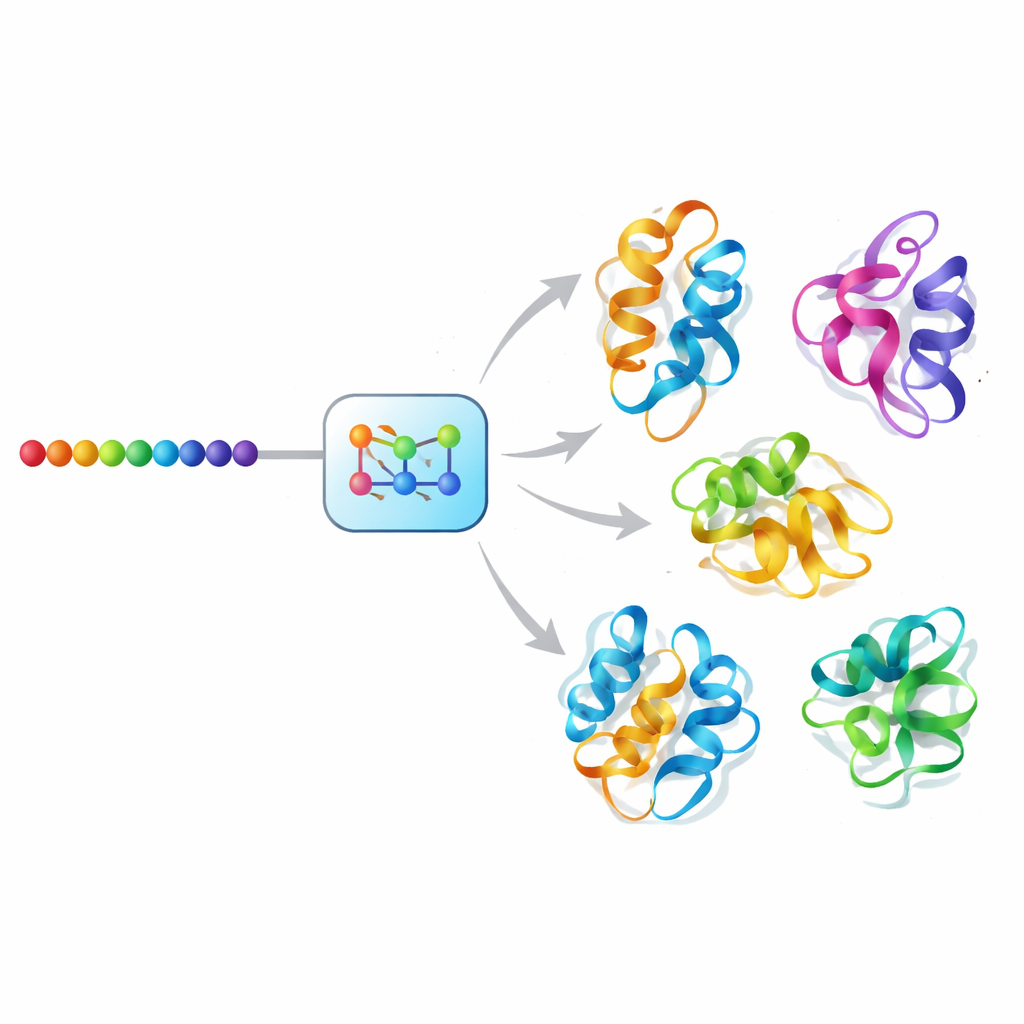
En AI-diffusionsmetod för proteinrörelse
Mac-Diff tar sig an denna utmaning med en diffusionsliknande generativ modell, en klass av AI som drivit nyliga framsteg inom bildsyntes. Istället för att ta bort brus från fotografier så tar Mac-Diff bort brus från abstrakta geometriska beskrivningar av proteinets ryggrad. Modellen representerar ett protein som ett rutnät av parvisa relationer mellan dess rester — avstånd och vinklar som är okänsliga för hur hela molekylen är roterad eller translaterad. I ett framåtskridande steg tillsätts successivt brus till dessa geometriska mönster tills de liknar slumpmässigt statiskt brus. I återställningssteget lär den sig att ta bort bruset steg för steg, vägledd av proteinets aminosyrasekvens, tills koherenta 3D-kompatibla geometrier framträder, vilka sedan kan omvandlas till fullständiga atommodeller med standardprogram för strukturbyggnad.
Låta sekvens och struktur prata lokalt
En central innovation ligger i hur Mac-Diff kopplar en linjär sekvens av rester till deras 3D-grannar. Att låta varje rest uppmärksamma varje annan rest, som i text-till-bild-modeller, skulle istället sudda ut viktiga fysikaliska begränsningar. Författarna introducerar därför en "lokalitetsmedveten" uppmärksamhetsmekanism som fokuserar varje rest på ett litet, sannolikt grannskap av interaktionspartners. För att uppskatta dessa grannskap använder Mac-Diff tre ingredienser: en protein-språkmodell kallad ESM-2 som kodar varje rests biokemiska kontext; en kontaktkarta som antyder vilka par av rester som sannolikt ligger nära varandra; och en enkel regel som gynnar rester som ligger nära varandra längs kedjan. Dessa signaler kombineras så att modellen under brusborttagningen föredrar information från rester som är fysiskt plausibla partners, vilket skärper dess förmåga att återskapa realistiska, flexibla strukturer.
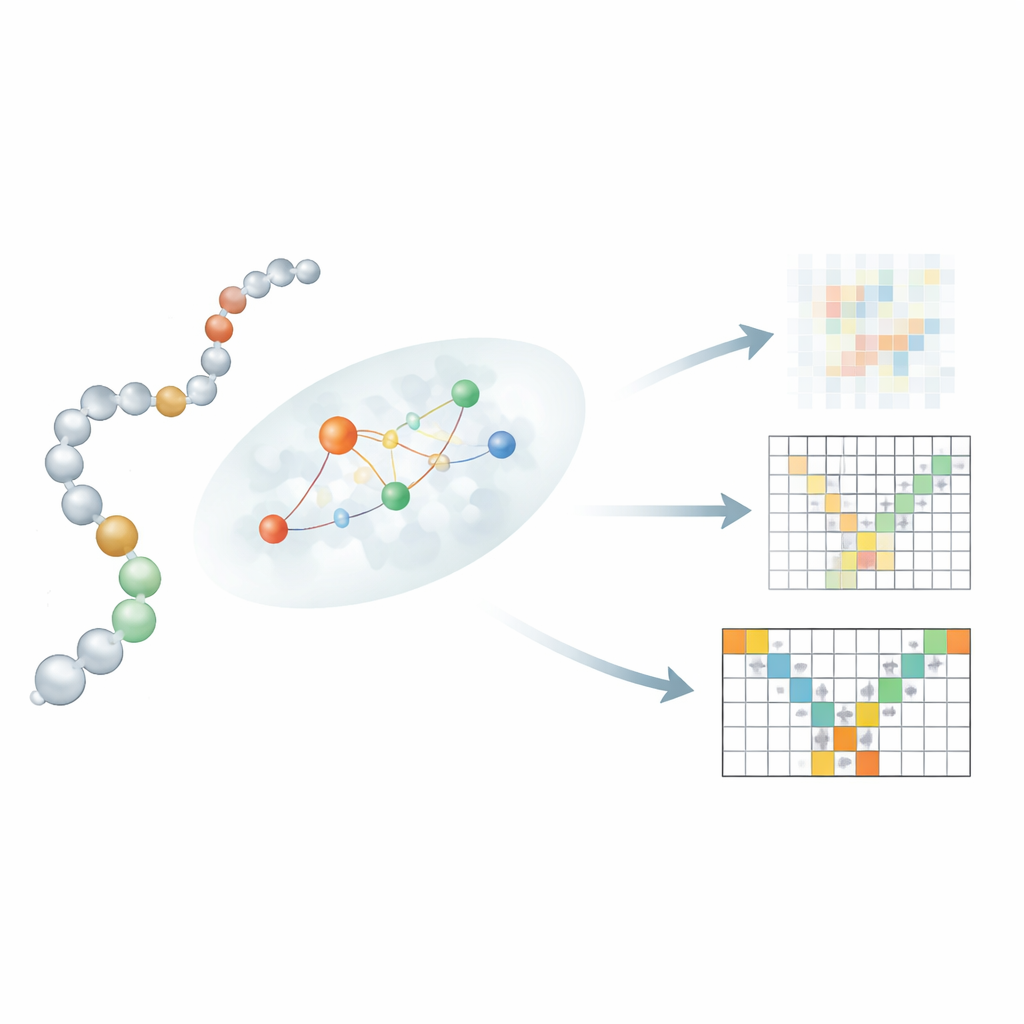
Testning mot långa simuleringar och formförändrande proteiner
Forskarlaget testade Mac-Diff på två krävande fronter. För det första undrade de om det kunde återskapa den breda fördelning av former som ses i långa, noggrant beräknade molekylärdynamiksimuleringar av snabbt veckande proteiner och ett klassiskt referensprotein känt som BPTI. Över flera mått som jämför statistiska egenskaper hos de genererade ensemblerna med simuleringsdata — såsom fördelningar av avstånd inom proteinet och övergripande kompakthet — matchade eller överträffade Mac-Diff konkurrerande AI-metoder, samtidigt som den genererade en större variation av konformationer. Den fångade de flesta av de viktiga "metastabila" tillstånden som identifierades i simuleringarna och återskapade rester-nivå flexibilitetsmönster med hög korrelation, vilket visar att dess ensembler speglar både globala vikningar och lokala rörelser på ett realistiskt sätt.
Avslöja dolda funktionella tillstånd
För det andra utmanade teamet Mac-Diff med proteiner som är kända för att anta mycket olika former i sitt arbete, inklusive enzymet adenylatkinas, som växlar mellan öppna och stängda former under energimetabolism, och en kurerad uppsättning av 40 proteiner med två experimentellt bestämda konformationer vardera. Mac-Diff genererade bara 100 kandidatstrukturer per protein — betydligt färre än typiska simuleringsbanor — men återfann ändå de flesta kända tillstånd med god geometrisk överensstämmelse. I adenylatkinas producerade den till exempel både öppna och stängda konformationer med hög likhet till kristallstrukturer, medan flera populära metoder tenderade att favorisera endast ett tillstånd. Modellen kördes också omkring tusen gånger snabbare än konventionella simuleringar på jämförbar hårdvara, vilket gör systematisk utforskning av formmångfald mycket mer praktisk.
Vad detta betyder för biologi och medicin
I vardagliga termer förvandlar Mac-Diff en proteins sekvens till ett galleri av rimliga poser snarare än ett enda porträtt, och den gör det med förståelse för vilka delar som sannolikt knuffar eller griper tag i varandra i 3D. Genom att noggrant och effektivt sampla dessa ensembler erbjuder metoden ett sätt att undersöka hur subtila formskiften ligger till grund för funktion, att upptäcka sällsynta men viktiga konformationer och att söka efter läkemedelsbindande fickor som endast uppträder i övergående tillstånd. Även om den ännu inte fångar de fullständiga tidssekvensfilmer som simuleringar kan ge, gör Mac-Diff det dynamiska landskapet hos proteiner tillgängligt för många fler system och lovar nya insikter inom strukturell biologi, läkemedelsdesign och proteinengineering.
Citering: Wang, B., Wang, C., Chen, J. et al. Conditional diffusion with locality-aware modal alignment for generating diverse protein conformational ensembles. Nat Mach Intell 8, 415–434 (2026). https://doi.org/10.1038/s42256-026-01198-9
Nyckelord: proteindynamik, diffusionsmodeller, konformationsensembler, allosteriska proteiner, läkemedelsupptäckt