Clear Sky Science · sv
Enastående robusthet hos fysikinformerade atomenergimodeller vid och över rumstemperatur
Varför detta är viktigt för vardaglig kemi
Datorsimuleringar är arbetsdjuren inom modern kemi och materialvetenskap. De låter forskare iaktta hur molekyler vrider sig, vibrerar och kolliderar in silico istället för i dyra, tidskrävande experiment. Men när dessa simuleringar förlitar sig på maskininlärning kan de plötsligt "explodera" och ge omöjliga molekylformer — särskilt vid högre temperaturer. Denna studie introducerar en ny typ av fysikinformerad maskininlärningsmodell som kan köra sådana simuleringar mycket länge, vid temperaturer upp till 1000 kelvin, utan att falla sönder.
Från smarta genvägar till sköra simuleringar
Traditionell kvantkemi beräknar molekylenergier med hög noggrannhet men är smärtsamt långsam. Enklare kraftfält är snabba men ofta approximativa. Maskininlärda potentialer strävar efter att kombinera det bästa av båda världar: de lär sig en genväg från molekylgeometri till energi och krafter, och använder sedan den genvägen för att driva molekylär dynamik. På papper ser många sådana modeller utmärkta ut, med mycket små genomsnittliga fel på standardtestset. I praktiken kan dessa siffror vara missvisande. När molekyler utforskar nya former under en simulering — särskilt vid högre temperaturer — pressas många modeller utanför det strukturrum de tränats på. Istället för att varsamt styra molekyler tillbaka till realistiska former kan de förutsäga krafter som töjer eller krossar bindningar tills hela systemet blir orealistiskt och simuleringen kraschar.
Att bygga modeller på kvantbaserade byggstenar
Författarna angriper denna skörhet genom att ändra vad modellen lär sig och hur den styrs av tidigare fysikaliska antaganden. De använder ett ramverk kallat FFLUX, som bygger på Interacting Quantum Atoms (IQA)-metoden. I IQA delas en molekyl in i "topologiska atomer" vars individuella energier bestäms direkt från kvantmekanik. Dessa atomenergier är fysiskt meningsfulla och summerar till molekylens totala energi. Istället för att lära sig godtyckliga platsenergier lär de nya Gaussiska processmodellerna dessa kvantrotade atomenergier, vilket ger ett djupt fysiskt ankare för varje prediktion. Fyra flexibla organiska molekyler — peptidkapslad glycin och serin, malondialdehyd och aspirin — fungerar som utmanande testfall på grund av sina många interna rörelser och den kända svårigheten för befintliga maskininlärda kraftfält.
Att lära modellen att förvänta sig problem
En viktig innovation ligger i hur den Gaussiska processen är uppbyggd innan den ens ser data: dess "medelfunktion", som kodar vad modellen antar i dåligt kända regioner. De flesta tidigare arbeten sätter helt enkelt detta medel till noll, vilket i praktiken låtsas att modellen inte har några förväntningar. Författarna flyttar istället medvetet detta medel mot atomenergier med högre värden, samtidigt som de håller det fysikaliskt rimligt. Detta designval innebär att när modellen tvingas extrapolera — till exempel när bindningar tillfälligt överspänns — föredrar den naturligt förutsägelser som straffar extrema distorsioner. I omfattande tester betedde sig modeller som endast skiljde sig i denna prior mycket olika. Modeller med konventionella eller lågenergimedel överlevde ofta mindre än en pikosekund innan en molekyl exploderade eller imploderade. I kontrast gav det bästa högenergimedlet (döpt MF5) simuleringar som förblev stabila i hela nanosekundstestfönstret vid temperaturer från 300 till 1000 kelvin för samtliga fyra molekyler.
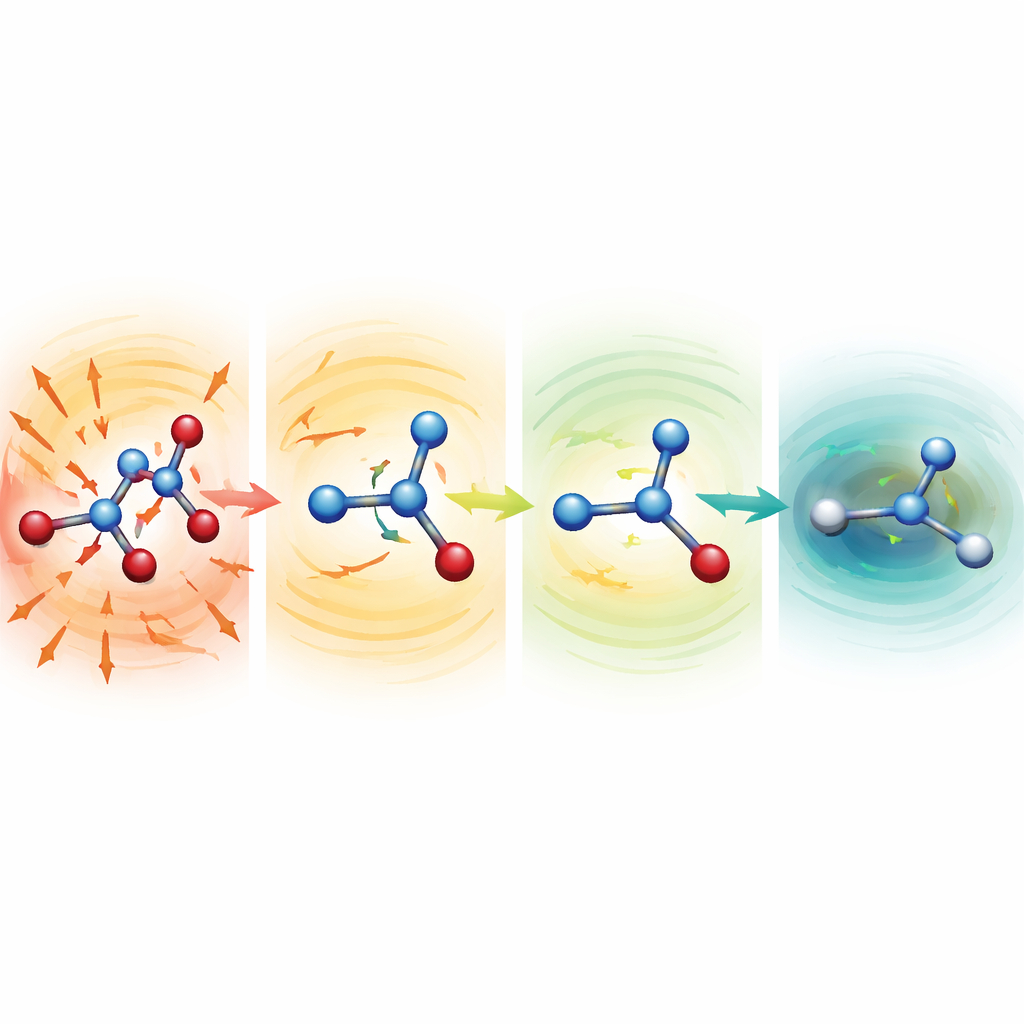
Att se förvrängda molekyler läka sig själva
För att undersöka varför de robusta modellerna fungerar så väl startade forskarna simuleringar från avsiktligt söndermanglade strukturer med kraftigt utdragna eller komprimerade bindningar. För serin, aspirin och malondialdehyd låg dessa startpunkter hundratals till mer än tusen kilokalorier per mol över en normal struktur — konfigurationer som normalt skulle vara katastrofala. Med svagare medelfunktioner flög molekylerna snabbt isär. Med MF5-upplägget pekade de predikterade krafterna omedelbart i riktningar som förkortade utdragna bindningar och förlängde tilltryckta. Inom tiotals till hundratals tidssteg relaxerade molekylerna till realistiska former och fortsatte sedan att utvecklas stabilt. Gruppen visade också att, utan att någonsin tränas på krafter, kan samma modeller styra geometrioptimeringar av alanindipeptid och återge kända lågenergikonformationer och relativa energier inom några tiondelar av en kilokalori per mol, men till ungefär 200 gånger lägre kostnad än fullständiga kvantberäkningar.
Långa, heta simuleringar på vanlig hårdvara
Robusthet handlar inte bara om att överleva en svår start; det handlar om att hålla över miljontals eller miljarder tidssteg. Författarna pressade sina bästa modeller ytterligare genom att köra 50 oberoende simuleringar vid 500 kelvin, vardera i 10 nanosekunder, för sina fyra testmolekyler. Ingen av dessa körningar kraschade, vilket gav en samlad simuleringstid på en halv mikrosekund — ovanligt för toppmoderna maskininlärda kraftfält. Ännu mer anmärkningsvärt var att simuleringarna kördes effektivt på vanliga CPU:er, steg-för-steg konkurrerande med eller överträffande vissa framstående neurala nätverkspotentialer som kräver kraftfulla GPU:er. Genomgående utforskade molekylerna rika uppsättningar former och metastabila tillstånd, vilket visar att robusthet inte uppnåddes genom att artificiellt frysa rörelse eller tvinga fram stela strukturer.
Vad detta betyder för framtida molekylmodellering
För icke-experter är huvudbudskapet att inte alla maskininlärningsmodeller med lågt fel är pålitliga när de pressas hårt. Genom att förankra sina modeller i kvantavledda atomenergier och noggrant ställa in modellens inbyggda förväntningar mot högenergistater skapade författarna en familj av potentialer som naturligt genererar "återställande krafter" — molekylära motsvarigheter till ett säkerhetssel — som håller simuleringar fysiska även vid höga temperaturer och från förvrängda startpunkter. Detta tillvägagångssätt lovar mer tillförlitliga och längre simuleringar av komplexa molekyler, och pekar mot framtida utvidgningar där liknande fysikinformerade modeller hanterar kondenserade faser och subtila interaktioner som dispersion, samtidigt som de förblir beräkningsmässigt praktiska.
Citering: Isamura, B.K., Aten, O., Nosratjoo, M. et al. Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature. Commun Chem 9, 138 (2026). https://doi.org/10.1038/s42004-026-01965-0
Nyckelord: maskininlärda kraftfält, molekylär dynamiks robusthet, Gaussiska processpotentialer, fysikinformerad modellering, kvantbaserade atomenergier