Clear Sky Science · sv
Jämförelse av djupinlärningsmodeller för att förutsäga anticancer-drogeffektivitet (IC50) med insikter för medicinalkemister
Varför denna forskning är viktig för framtida cancerläkemedel
Att designa nya cancerläkemedel är långsamt och kostsamt eftersom varje lovande molekyl måste testas i levande celler för att avgöra hur starkt den hämmar deras tillväxt. Denna studie ställer en praktisk fråga: kan moderna artificiella intelligensverktyg på ett tillförlitligt sätt förutsäga dessa testresultat i förväg och därigenom spara tid och kostnad i laboratoriet? Författarna jämför systematiskt flera populära djupinlärningssystem, undersöker när de lyckas eller misslyckas, och föreslår till och med ett mer realistiskt sätt att bedöma deras användbarhet för verksamma medicinalkemister.
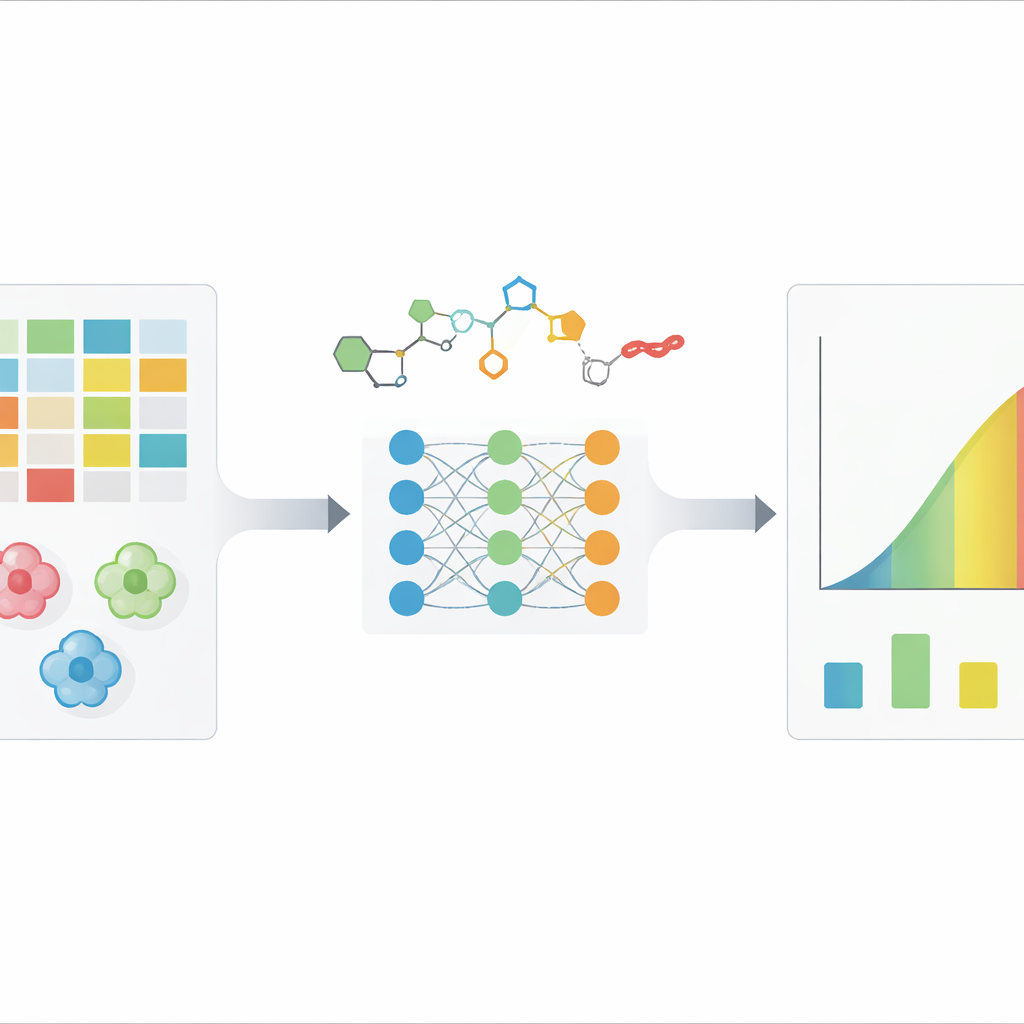
Mätning av hur starkt ett läkemedel kämpar mot cancerceller
När forskare testar en potentiell anticancerförening rapporterar de ofta ett tal som kallas IC50: den koncentration vid vilken läkemedlet reducerar celltillväxten till hälften. Ett lågt IC50 innebär ett potent läkemedel. Men samma förening kan ha mycket olika IC50-värden i olika cancercellinjer, och även upprepade tester av samma läkemedel–cellpar kan variera flera gånger beroende på assay och förhållanden. Traditionella metoder för datorstödd design fångar hur en molekyl passar en enskild proteintarget, men har svårt att hantera den fulla komplexiteten i levande celler. Nyare djupinlärningsmetoder försöker lära sig mönster direkt från stora dataset som länkar kemiska strukturer och detaljerad genetisk information om cancerceller till deras uppmätta IC50-värden.
Sätta fem djupinlärningsverktyg på prov
Författarna undersökte fem ledande djupinlärningsmodeller, var och en med en annan strategi för att representera både läkemedel och cancerceller. Vissa behandlar molekyler som grafer av atomer och bindningar; andra omvandlar cellgenetik till strukturerade nätverk av biologiska processer eller lyfter fram de mest informativa generna. Alla modeller tränades och utvärderades på samma kurerade data från en större resurs kallad GDSC, som innehåller tiotusentals uppmätta IC50-värden. Teamet byggde också en medvetet enkel jämförelsemetod: en "baseline" som ignorerar biologi och kemi och bara förutsäger genomsnittliga IC50-värden från träningsdata. Detta gjorde det möjligt att ställa frågan inte bara vilken djupmodell som är bäst, utan om någon av dem verkligen överträffar en mycket naiv genväg.
Ett mer realistiskt sätt att poängsätta förutsägelser
Vanliga maskininlärningsmått, såsom korrelation och root-mean-squared error, kan se imponerande ut men är svåra för laboratorieforskare att tolka. För att överbrygga detta gap omformulerade författarna förutsägelsekvalitet i mer intuitiva termer, såsom procentuell felmarginal och fel på en logaritmisk skala som motsvarar viktskillnader i IC50. Avgörande var också att de kvantifierade hur brusiga verkliga IC50-mätningar är genom att gräva i en stor bioaktivitetsdatabas. De visade att, under vanliga assayförhållanden, hamnar 90 % av upprepade IC50-mätningar för samma läkemedel–cellpar inom ungefär en sju-faldig intervall. Med detta definierade de en ny metrisk, Experimental Variability-Aware Prediction Accuracy (EVAPA): andelen modellförutsägelser som hamnar inom detta experimentellt realistiska band.
Var modellerna briljerar och var de kämpar
När data slumpmässigt delades så att många läkemedel och cellinjer förekom i både tränings- och testset presterade alla djupinlärningsmodeller väl. De visade stark korrelation med uppmätta IC50-värden och höga EVAPA-poäng, och slog tydligt den enkla baslinjen. Prestationen hölls relativt god när modellerna ombads generalisera till helt nya cellinjer samtidigt som de fortfarande såg bekanta läkemedel; i detta fall gjorde även baslinjen oväntat bra ifrån sig, vilket tyder på att genomsnittligt läkemedelsbeteende över många celltyper redan bär användbar information. Det verkliga problemet uppstod när modellerna mötte nya kemiska strukturer: noggrannheten sjönk kraftigt, korrelationerna närmade sig noll eller blev till och med negativa, och i vissa tester matchade eller överträffade den enkla baslinjen de djupa modellerna. Teamet undersökte också om förutsägelsefelen berodde på grundläggande läkemedelsegenskaper såsom storlek, polaritet eller flexibilitet, eller på vävnadsursprunget för cellinjerna. De fann endast svaga samband, vilket antyder att modellerna fungerar någorlunda lika över olika kemier och cancertyper — men fortfarande sviktar för verkligt nya föreningar.
Pröva verkligen nya molekyler från nyare studier
För att gå bortom offentliga databaser samlade författarna mer än 150 nyligen rapporterade anticancerföreningar från medicinalkemisk litteratur och testade flera av djupinlärningsmodellerna på dessa otestade molekyler. Resultaten speglade "nytt läkemedel"-scenariot i GDSC-data: förutsägelserna var brusiga, med stora procentuella fel och endast måttliga andelar förutsägelser inom realistiska experimentella gränser. Ändå antydde modellernas beteende över olika assaytyper att de fångade vissa assayoberoende mönster i hur läkemedel påverkar celler. En enkel webbserver byggd från dessa modeller låter nu kemister mata in en struktur och få förutsagda IC50-värden för hundratals cancercellinjer, med förbehållet att tillförlitligheten är högst när den nya molekylen liknar dem som redan finns i träningssetet.
Vad detta betyder för läkemedelsupptäckt
Detta arbete visar att nuvarande djupinlärningsverktyg redan är användbara för att rangordna och utforska idéer för cancerläkemedel när de verkar inom bekant kemiskt territorium, men att de är långt ifrån att vara kristallkulor för verkligt nya molekylära designer. Genom att lyfta fram att en grov genomsnittsbaserad modell ibland kan väga upp mot komplexa neurala nätverk, och genom att introducera ett noggrannhetsmått förankrat i verklig experimentell variabilitet, ger studien medicinalkemister en tydligare bild av vad de kan förvänta sig av IC50-förutsägelseprogramvara. Budskapet är avvägt: dessa modeller är lovande hjälpmedel för läkemedelsupptäckt, särskilt när de noggrant jämförs, men betydande framsteg i arkitektur och träning — särskilt för molekyler utanför träningsfördelningen — behövs fortfarande innan de pålitligt kan vägleda sökandet efter nästa generations cancerterapier.
Citering: Garai, U., Pal, A.S., Ghosh, K. et al. Benchmarking deep learning models for predicting anticancer drug potency (IC50) with insights for medicinal chemists. Commun Chem 9, 106 (2026). https://doi.org/10.1038/s42004-026-01916-9
Nyckelord: anticancer-drogeffektivitet, IC50-förutsägelse, djupinlärningsmodeller, cancercellinjer, beräkningsbaserad läkemedelsupptäckt