Clear Sky Science · sv
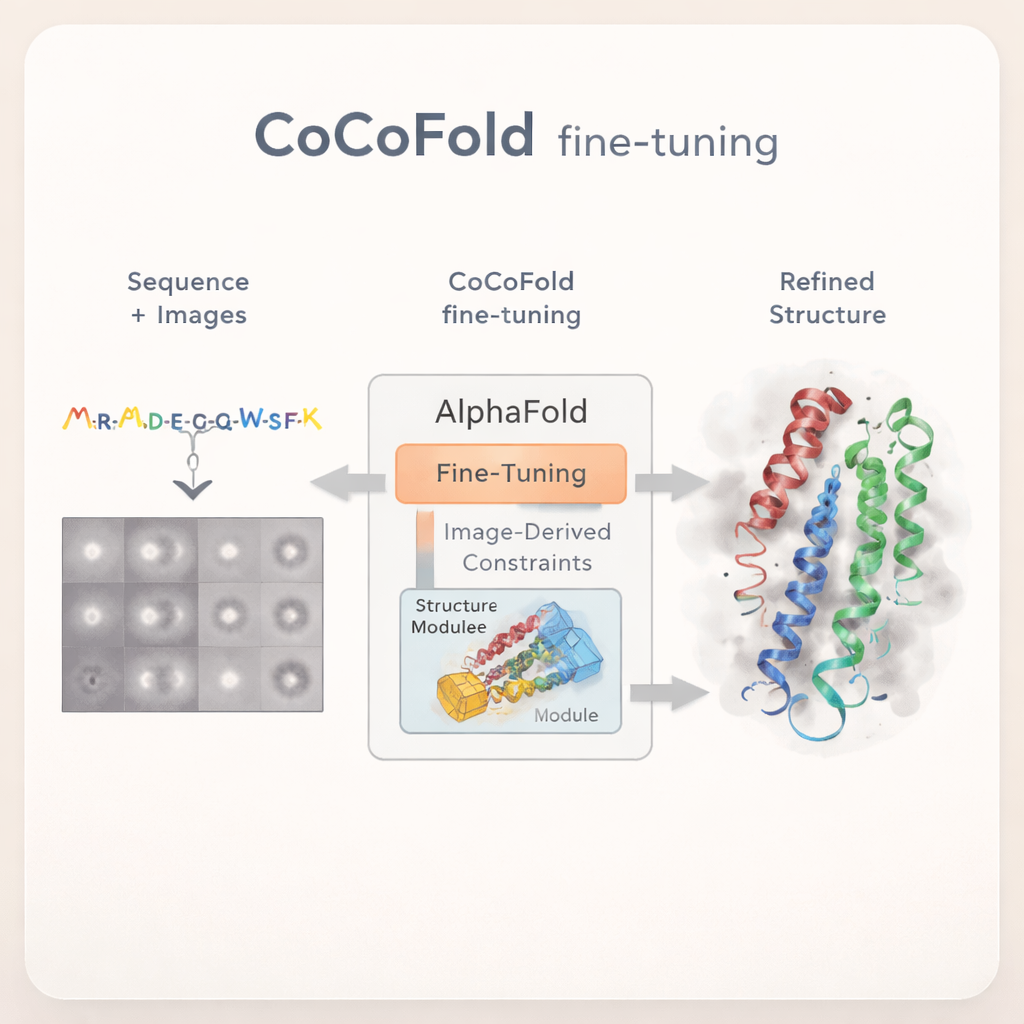
Finjustering av AlphaFold med begränsade cryo-EM-observationer
Varför proteiners former är så svåra att se
Proteiner är små molekylära maskiner som driver nästan alla processer i våra kroppar, från energiproduktion till nervsignaler. För att förstå hur de fungerar — och hur läkemedel kan påverka dem — behöver forskare deras precisa tredimensionella former. Två kraftfulla verktyg har vuxit fram för detta: cryo‑elektronmikroskopi (cryo‑EM), som tar många suddiga ögonblicksbilder av frysta proteiner, och AlphaFold, ett artificiellt intelligenssystem som förutsäger proteinstrukturer från deras sekvenser. Men i många verkliga experiment är cryo‑EM‑data ofullständiga, och AlphaFolds prediktioner stämmer inte alltid med verkligheten. Denna artikel presenterar CoCoFold, en metod som lär AlphaFold att lyssna direkt på svåra cryo‑EM‑data och förbättra sina förutsägelser därefter.
När kameran ser för lite
Cryo‑EM fungerar genom att snabbt frysa proteiner och avbilda enorma mängder enskilda partiklar från många vinklar, för att sedan kombinera dessa bilder till en 3D‑karta. I praktiken har forskare dock ofta inte tillräckligt många bra bilder att arbeta med. Ibland förekommer proteinet bara kort i ett högenergitillstånd, så väldigt få partiklar fångas. I andra fall föredrar proteiner vissa orienteringar på isytan, vilket gör att många siktvinklar saknas. Båda problemen leder till oskarpa, ofullständiga kartor som är svåra att översätta till tillförlitliga atommodeller. Befintlig programvara kan passa AlphaFolds förutsagda strukturer in i sådana kartor, men framgången är starkt beroende av att man börjar med skarpa, högupplösta data.
Att lära AlphaFold att läsa råa bilder
CoCoFold tar ett annat grepp: istället för att förlita sig på en fullständigt rekonstruerad 3D cryo‑EM‑karta använder den direkt de råa 2D‑partikelbilderna för att finjustera AlphaFold. Metoden utgår från en AlphaFold‑Multimer‑prediktion och håller större delen av det ursprungliga nätverket fryst, så att dess breda kunskap om proteinveckning bevaras. Endast den sista struktur‑byggande delen tillåts ändras. En lättviktig ”adapter” läggs till för att mata in information härledd från cryo‑EM‑bilderna till strukturmodulen, och på så vis vägleder modellen mot former som är förenliga med de experimentella uppgifterna samtidigt som den undviker vilda avvikelser från känd proteinfysik.
Att omvandla bilder till strukturell återkoppling
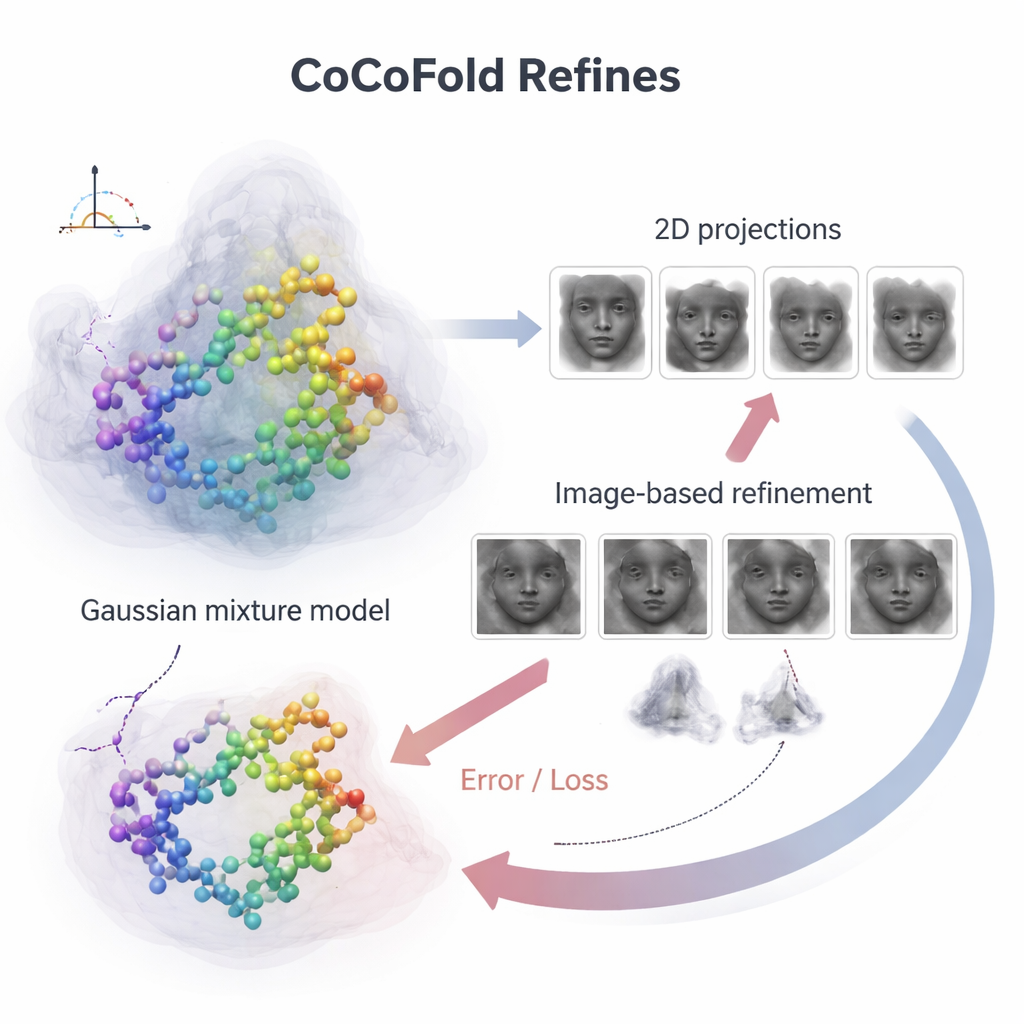
För att koppla enskilda proteinatomer till de brusiga mikroskopbilderna bygger CoCoFold en mjuk, flexibel bild av den förutsagda strukturen med överlappande tredimensionella klumpar, kända som en Gaussisk blandning. Från denna representation simulerar den hur proteinet skulle se ut i mikroskopet vid samma vyer och avbildningsförhållanden som i det verkliga experimentet. Dessa simulerade ögonblicksbilder jämförs sedan med de faktiska cryo‑EM‑partiklarna, ring för ring i frekvensdomänen, för att se hur väl de stämmer. Varje missanpassning blir en återkopplingssignal som flyter tillbaka genom nätverket och justerar både proteinmodellen och densitetsrepresentationen en aning. Efter träning städas atommodellen ytterligare upp med ett fysikbaserat förfiningssteg för att ta bort lokala geometriska kollisioner.
Att hålla noggrannheten när data är knappa eller snedvridna
Författarna testade CoCoFold på flera experimentella och simulerade dataset utformade för att efterlikna de två huvudproblemen i cryo‑EM: för få partiklar och stora luckor i siktvinklar. Under dessa svåra förhållanden tenderade standardverktyg — inklusive andra djupinlärningsmetoder som är beroende av rekonstruerade kartor — att missa delar av proteinet, förflytta helixar eller tappa finare detaljer i takt med att kartorna blev suddigare. CoCoFold, däremot, producerade konsekvent modeller som överensstämde bättre och mer fullständigt med kända referensstrukturer. Dess fel förblev små även när antalet partiklar minskades drastiskt eller när stora koner av siktvinklar saknades, vilket tyder på att direktinlärning från råbilder bevarar viktig information som kartbaserade angreppssätt kastar bort.
Vad detta betyder för framtidens strukturella biologi
För icke‑specialister är huvudbudskapet att CoCoFold fungerar som en översättare mellan kraftfulla AI‑förutsägelser och ofullkomliga experimentella data. Istället för att lita enbart på AlphaFold eller cryo‑EM låter den de två informera varandra, särskilt i svåra lägen där experimenten bara ger en partiell bild. I enkla fall med rikliga, högkvalitativa data fungerar fortfarande befintliga kartstyrda verktyg mycket väl. Men när partiklar är sällsynta eller orienteringar saknas — vanliga situationer vid studier av flyktiga eller bräckliga proteintillstånd — erbjuder CoCoFold ett sätt att återskapa tillförlitliga atommodeller från information som annars skulle gå förlorad.
Citering: Liao, J., Zheng, D., Zhang, H. et al. Fine-tuning AlphaFold with limited cryo-EM observations. Commun Chem 9, 95 (2026). https://doi.org/10.1038/s42004-026-01899-7
Nyckelord: cryo-EM, AlphaFold, proteinstruktur, djupinlärning, strukturell biologi