Clear Sky Science · sv
Histondemetylas KDM7A reglerar negativt profibrotisk makrofagpolarisering och lungfibrosprogression
Varför ärrbildning i lungorna berör oss alla
När lungorna utvecklar envisa ärr blir andningen en daglig kamp. Detta tillstånd, känt som pulmonell fibros, drabbar miljontals och saknar i dag botemedel—det finns endast läkemedel som bromsar skadorna. I denna studie upptäcker forskare en tidigare dold molekylär ”broms” inne i immunceller kallade makrofager som hjälper till att hålla lungärrbildning i schack. Att förstå denna broms kan öppna dörren för nya behandlingar, inte bara för lungfibros utan potentiellt även för andra sjukdomar där skadlig ärrbildning och okontrollerad inflammation går hand i hand.
En berättelse om formbytande immunceller
Makrofager är immunsystemets frontlinjeceller som patrullerar vävnader, rensar bort skräp och hjälper till att reparera skador. Men de är också formbytare: i vissa situationer blir de proinflammatoriska krigare, medan de i andra förvandlas till sårläkare som kan driva ärrbildning. En särskild ärrfrämjande typ, kallad profibrotiska makrofager (Fib-Mac), är starkt kopplad till lungfibros. Dessa celler producerar molekyler som aktiverar fibroblaster, vilka i sin tur lägger ned överdriven mängd kollagen och andra matrixkomponenter, och successivt styvar upp lungan. Författarna ville ta reda på hur de genetiska ”inställningarna” inne i makrofager avgör om de blir dessa farliga Fib-Mac-celler eller förblir i mer balanserade, skyddande tillstånd.

En epigenetisk broms gömd i genomet
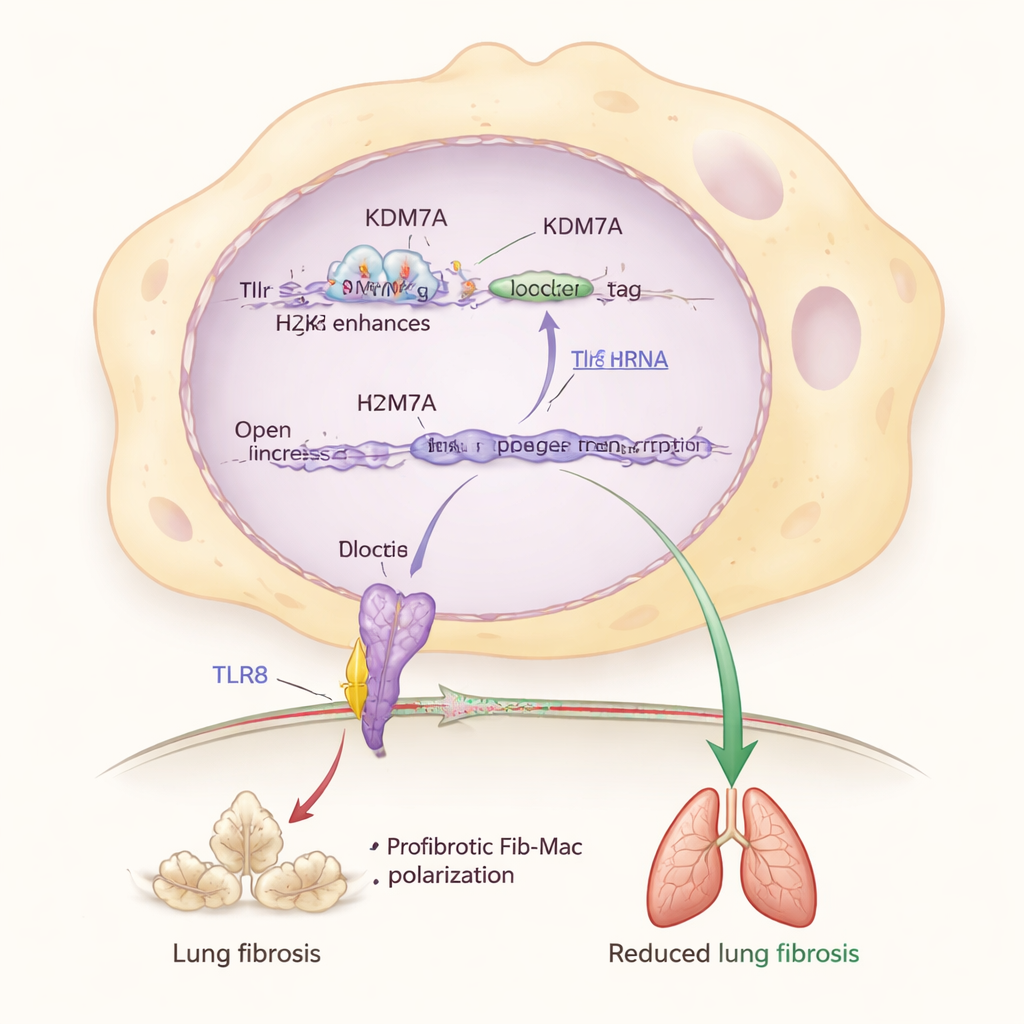
Teamet började med att skanna hundratals kända epigenetiska regulatorer—proteiner som finjusterar hur tätt DNA paketeras och vilka gener som slås på eller av. Genom att använda RNA-sekvensering i både mänskliga och musmakrofager fann de att ett enzym kallat KDM7A starkt slås på när makrofager pressas mot ett fibrotiskt, sårläkande tillstånd. KDM7A är en "histondemetylas": det tar bort vissa kemiska märkningar från histonproteiner runt vilka DNA är lindat. Det mönstret antydde att KDM7A kan fungera som en inbyggd återkopplingsbroms, aktiverad just när makrofager börjar driva mot en ärrfrämjande identitet.
För att testa detta använde forskarna möss som saknar genen Kdm7a och orsakade lungskada med cytostatikumet bleomycin, en standardmodell för pulmonell fibros. Tidigt efter skadan såg lungvävnaden liknande ut i normala och Kdm7a‑deficienta djur. Men efter tre veckor visade möss utan Kdm7a mycket mer omfattande ärrbildning, kollaps av de små luftblåsorna och högre "Ashcroft‑poäng" som kvantifierar fibros. Gener involverade i kollagenproduktion och andra fibrosrelaterade banor var mer aktiva i dessa knockout‑möss, vilket bekräftar att förlust av Kdm7a gör lungorna mer sårbara för långvarig, skadlig ärrbildning.
Hur KDM7A styr makrofager bort från en ärrfrämjande öde
Med hjälp av enkelcells‑RNA‑sekvensering zoomade författarna in på individuella lungceller från skadade möss. De upptäckte att i frånvaro av Kdm7a expanderade en särskild makrofag‑undergrupp i lungans stödjevävnad dramatiskt och fick en stark Fib‑Mac‑signatur, med uttryck av gener som Arg1, Spp1 och Trem2. Ytterligare experiment i odlade makrofager visade att borttagning av Kdm7a ökade uttrycket av Fib‑Mac‑markörgener och omprogrammerade cellmetabolismen mot banor som stöder kollagenproduktion och varaktig aktivering. Med andra ord, KDM7A dämpar normalt både de genetiska och metaboliska programmen som driver makrofager in i ett fibrosfrämjande tillstånd.
När de grävde djupare identifierade forskarna en nyckelpartner i detta bromssystem: ett sensorprotein kallat TLR8, som upptäcker RNA‑bitar inne i immunceller. De fann att KDM7A hjälper till att hålla genen Tlr8 aktiv genom att ta bort ett repressivt kemiskt märke (H3K27me2) från en enhancerregion nära Tlr8. När Kdm7a var inaktiverad ackumulerades detta märke, Tlr8‑nivåerna föll och Fib‑Mac‑egenskaperna förstärktes. Direkt nedreglering av Tlr8 i makrofager pressade dem också mot en fibrotisk identitet, medan aktivering eller överproduktion av TLR8 drog tillbaka dem, även när Kdm7a saknades. Detta placerar KDM7A–TLR8‑vägen i centrum för en molekylär krets som skyddar lungorna från överdriven ärrbildning.
Från åldrande lungor till mänsklig sjukdom
För att koppla dessa fynd till människor undersökte teamet lungvävnad från patienter med fibrotisk lungsjukdom. Jämfört med icke‑sjuk vävnad innehöll fibrotiska lungor många fler makrofager med Fib‑Mac‑markörer, men dessa samma celler visade markant reducerade nivåer av KDM7A och TLR8. Omsanalys av befintliga enkelcellsdataset från patienter med idiopatisk pulmonell fibros bekräftade detta mönster: när Fib‑Mac‑signaturerna ökade föll KDM7A‑uttrycket. Forskarna grävde också i en stor musatlas och fann att Kdm7a‑ och Tlr8‑uttrycket i makrofager minskade med åldern hos hanar, vilket speglar den högre risken för pulmonell fibros hos äldre män. Detta tyder på att ålders‑ och könsrelaterad försvagning av KDM7A–TLR8‑bromsen kan bidra till att förklara vem som är mest sårbar för svår lungärrbildning.
Vad detta betyder för framtida behandlingar
Enkelt uttryckt visar detta arbete att vårt immunförsvar bär på en intern säkerhetsmekanism som förhindrar att hjälpsamma reparationsceller blir överivriga och förvandlas till drivkrafter för permanenta ärr. KDM7A, verksamt via TLR8, hindrar makrofager från att låsa sig i ett profibrotiskt läge och hjälper därigenom till att upprätthålla flexibel, funktionell lungvävnad efter skada. När detta system sviktar—genom genetisk förlust, åldrande eller andra faktorer—är makrofager mer benägna att bli "ärrförstärkare" som förvärrar fibros. Genom att avslöja denna epigenetiska broms pekar studien mot nya terapeutiska strategier: läkemedel som ökar KDM7A‑aktivitet, efterliknar dess effekter eller noggrant stimulerar TLR8 kan en dag komplettera befintliga antifibrotiska behandlingar och erbjuda bättre skydd mot progressiv, livsbegränsande lungärrbildning.
Citering: Funagura, N., Koga, T., Etoh, K. et al. Histone demethylase KDM7A negatively regulates fibrotic macrophage polarization and lung fibrosis progression. Commun Biol 9, 309 (2026). https://doi.org/10.1038/s42003-026-09610-1
Nyckelord: lungsfibros, makrofager, epigenetik, KDM7A, TLR8