Clear Sky Science · sv
haCCA: multi-module Integration of spot-based spatial transcriptomes and metabolomes
Varför det spelar roll att kartlägga molekyler på plats
Våra kroppar är uppbyggda av otaliga små cellområden, var och en med sin egen blandning av aktiva gener och kemiska substanser. Fram till nyligen tvingades forskare studera dessa molekyler först efter att vävnad mals till en homogen massa, vilket gör att all ”var” förloras. Den här artikeln presenterar en ny beräkningsmetod, kallad haCCA, som fogar samman två kraftfulla avbildningstekniker så att forskare in situ kan se hur gener och små molekyler är ordnade i verkliga vävnader och tumörer. En sådan karta kan avslöja dolda sjukdomsmönster och föreslå mer precisa behandlingar.
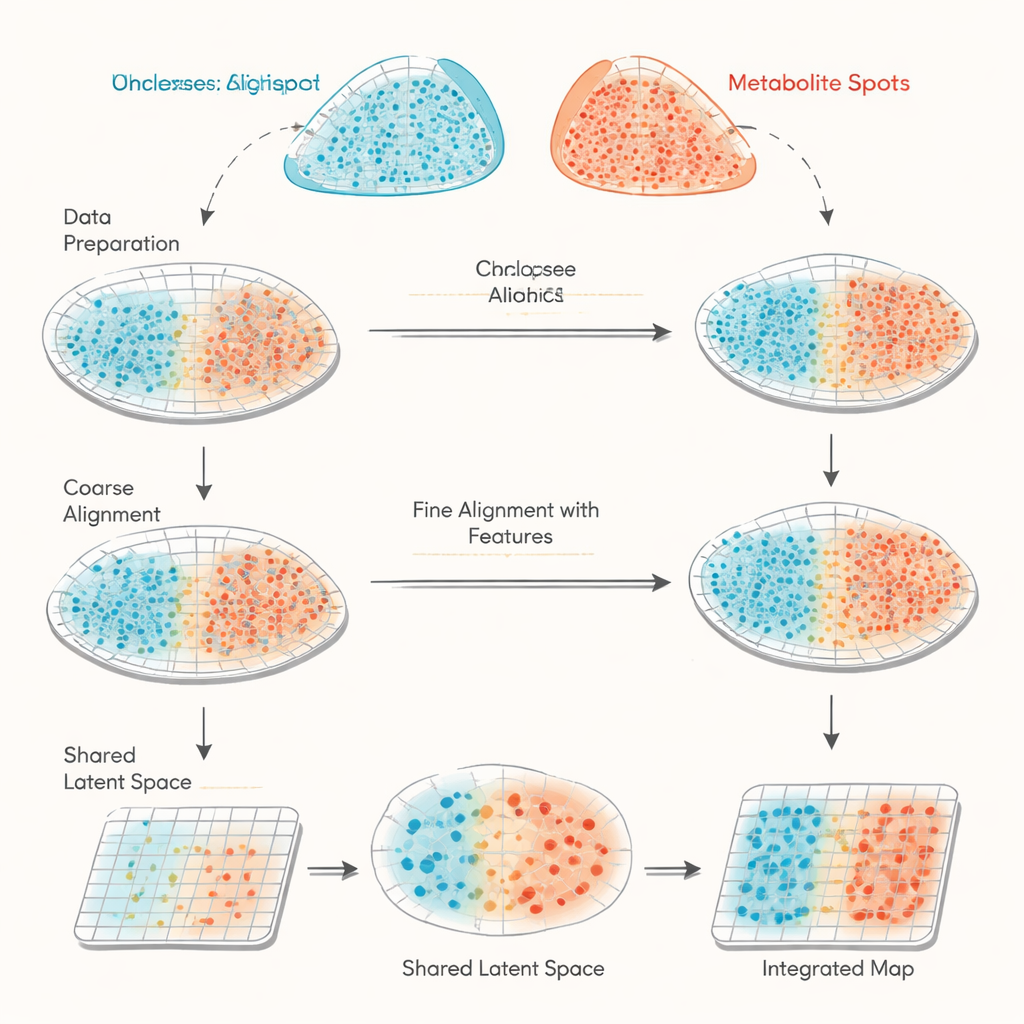
Två olika vyer av samma vävnad
Studien fokuserar på att kombinera data från två rumsliga metoder som blir allt vanligare inom biologi. Spatial transcriptomics registrerar vilka gener som är påslagna vid tusentals små punkter över en vävnadssnitt. MALDI-masspektrometriavbildning registrerar mängder av många små molekyler, såsom metaboliter och lipider, på likartat täta rutnät av punkter. Problemet är att dessa två instrument inte mäter exakt samma positioner eller samma uppsättning egenskaper, så deras data liknar två feljusterade kartor med olika förklaringsnycklar. Befintliga metoder försöker mest matcha vävnadssektionernas former enbart utifrån koordinater, vilket kan vara osäkert och sakna ett sätt att kontrollera hur väl justeringen faktiskt fungerade.
En smartare metod för att rada upp molekylkartor
haCCA (kort för hierarchical anchor-guided canonical correlation analysis) tar sig an denna utmaning genom att kombinera geometri med biologi. Först utförs en tvåstegs “morfologisk inpassning” av punktgridden från de två teknikerna. Människor med expertkunskap markerar några matchande landmärken på vävnadsbilderna för att grovt korrigera för förskjutningar och rotationer, och sedan finjusterar ett automatiserat steg avvikare nära trasiga kanter eller saknade delar. Därefter söker metoden efter ”ankare” — par av punkter som ligger nära varandra i rummet och befinner sig i lokalt homogena regioner, vilket gör dem sannolika representanter för samma vävnadsområde. Från dessa ankarpunkter beräknar haCCA vilka gener och metaboliter som tenderar att variera tillsammans och destillerar dem till en gemensam lågdimensionell representation som fångar deras starkaste gemensamma mönster.
Att omvandla korrelationer till en enhetlig vävnadsbild
Med både rumsliga koordinater och den delade molekylära representationen i handen löser haCCA ett optimeringsproblem för att avgöra hur sannolikt det är att varje genpunkt bör paras med varje metabolitpunkt. Detta steg är utformat för att hålla punkter nära i rummet men också lika i deras kombinerade gen–metabolitprofil. Slutresultatet är en ”transportplan” som länkar varje punkt i en datamängd till dess bästa partner i den andra, vilket skapar en integrerad multimodal karta. På noggrant konstruerade testdata—där de verkliga relationerna är kända—visar författarna att varje steg i arbetsflödet (grov inpassning, förfinad inpassning och funktionsmedveten matchning) stadigt förbättrar tre oberoende mått på noggrannhet. Jämfört med andra verktyg som huvudsakligen förlitar sig på geometri uppnår haCCA konsekvent bättre inpassning och en mer trogen överföring av regionsetiketter.
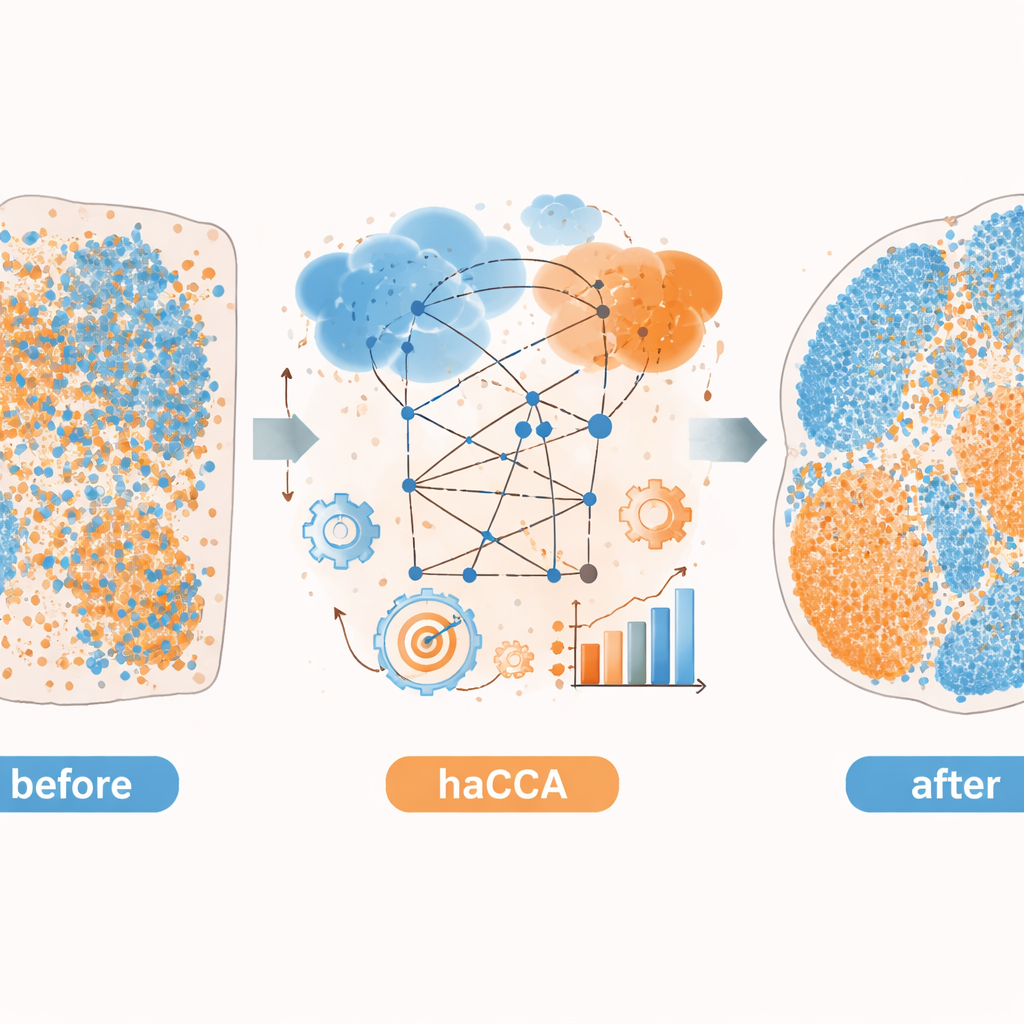
Avslöjar dold biologi i hjärn- och levercancer
Författarna tillämpar sedan haCCA på verkliga mus-hjärn- och levertumörvävnader. För hjärnan integrerar de kommersiella spatial transcriptomics-data med metabolitbilder från samma eller närliggande sektioner. Metoden bevarar kända metabola territorier och rekonstruerar förväntade överlappningar, såsom samexistensen av dopamin med genen som kodar för dess nyckelenzym. Genom att tillsammans klustra gener och metaboliter finner de att de kombinerade data avslöjar mer nyanserade vävnadsunderdelningar än någon av modaliteterna ensam. I en preklinisk modell av intrahepatisk kolangiocarcinom, en typ av levercancer, använder de haCCA för att jämföra tumörer som kan eller inte kan bilda neutrofilernas extracellulära nätverk—trådliknande strukturer som släpps ut av immunceller. De integrerade kartorna visar att när dessa nätverk är närvarande är en gen kallad Scd1 och dess associerade fettsyror förhöjda i maligna regioner, vilket pekar på en förskjutning mot förändrad fettmetabolism i tumören.
Vad detta innebär för framtida forskning
I vardagliga termer är haCCA som att rada upp flygfoton tagna med olika kameror—den ena känslig för byggnadsomriss, den andra för värmesignaturer—för att få en skarpare bild av vad som händer i varje stadsblock. Genom att noggrant slå samman var gener är aktiva med var nyckelmetaboliter ackumuleras hjälper detta arbetsflöde forskare att samtidigt profila båda sidorna av cellulärt beteende: instruktionerna och den resulterande kemin. Metoden förbättrar tidigare inpassningsmetoder, paketeras i ett lättillgängligt Python-verktyg och kan utvidgas till andra rumsliga teknologier. När sådana integrerade kartor blir mer rutinmässiga kan de fördjupa vår förståelse för hur tumörer och andra vävnader organiserar sin metabolism, svarar på behandlingar och utvecklas över tid.
Citering: Xu, J., Shen, XT., Zhang, C. et al. haCCA: multi-module Integration of spot-based spatial transcriptomes and metabolomes. Commun Biol 9, 248 (2026). https://doi.org/10.1038/s42003-026-09526-w
Nyckelord: spatial multi-omics, transcriptomics, metabolomics, tumor metabolism, data integration