Clear Sky Science · sv
Tolkbar maskininlärning förklarar hämning av karbonanhydras genom konform och kontrafaktisk prediktion
Varför smartare cancerläkemedel spelar roll
Cancerläkemedel fungerar ofta som trubbiga verktyg: samtidigt som de angriper tumörceller kan de också skada friska vävnader och ge allvarliga biverkningar. Ett lovande sätt att öka träffsäkerheten är att blockera specifika varianter av ett enzym kallat karbonanhydras, vilket hjälper tumörer att överleva i syrefattiga miljöer. Flera varianter av detta enzym ser dock nästan identiska ut, vilket gör det svårt att utforma läkemedel som träffar de ”skadliga” varianterna i tumörer utan att rubba den ”goda” som finns i hela kroppen. Denna studie visar hur tolkbar maskininlärning kan hjälpa forskare att navigera denna utmaning och designa mer selektiva, säkrare läkemedelskandidater.
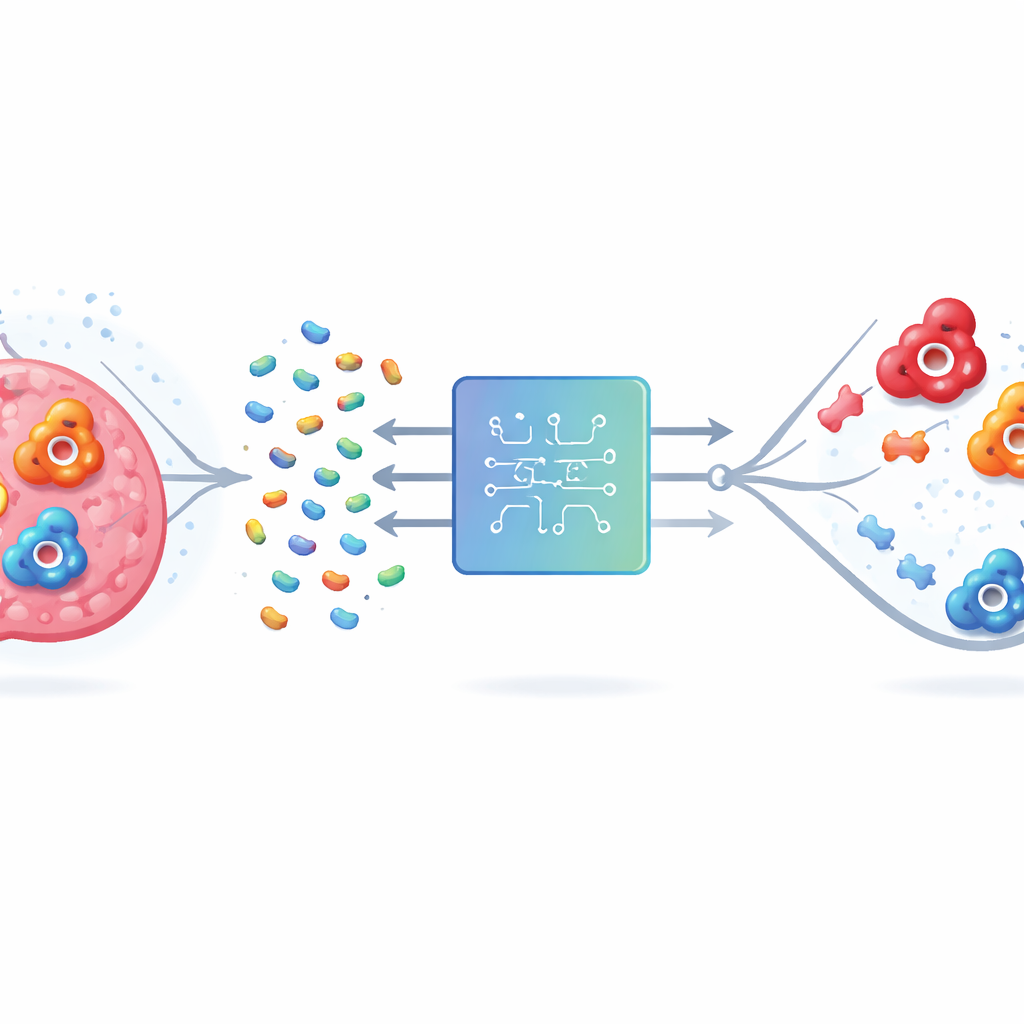
Problemet med att träffa fel mål
Mänsklig karbonanhydras (hCA) förekommer i många former, eller isoformer. Två av dem, IX och XII, är kopplade till cancercellers överlevnad i syrebrist i tumörer, så att blockera dem kan bromsa sjukdomen och förbättra behandling. Men isoform II är utbredd i friska vävnader och har ett aktivt centrum som liknar IX och XII mycket. Läkemedel som binder alla tre kan orsaka oönskade problem som metabol acidos och synrubbningar. Traditionella laboratorie- och datorbaserade metoder har svårt eftersom enzymer är stora, komplexa molekyler och antalet möjliga läkemedelslika föreningar är astronomiskt stort. Att testa dem alla uttömmande, vare sig i labbet eller på datorn, är helt enkelt inte genomförbart.
Bygga en ren och pålitlig databasis
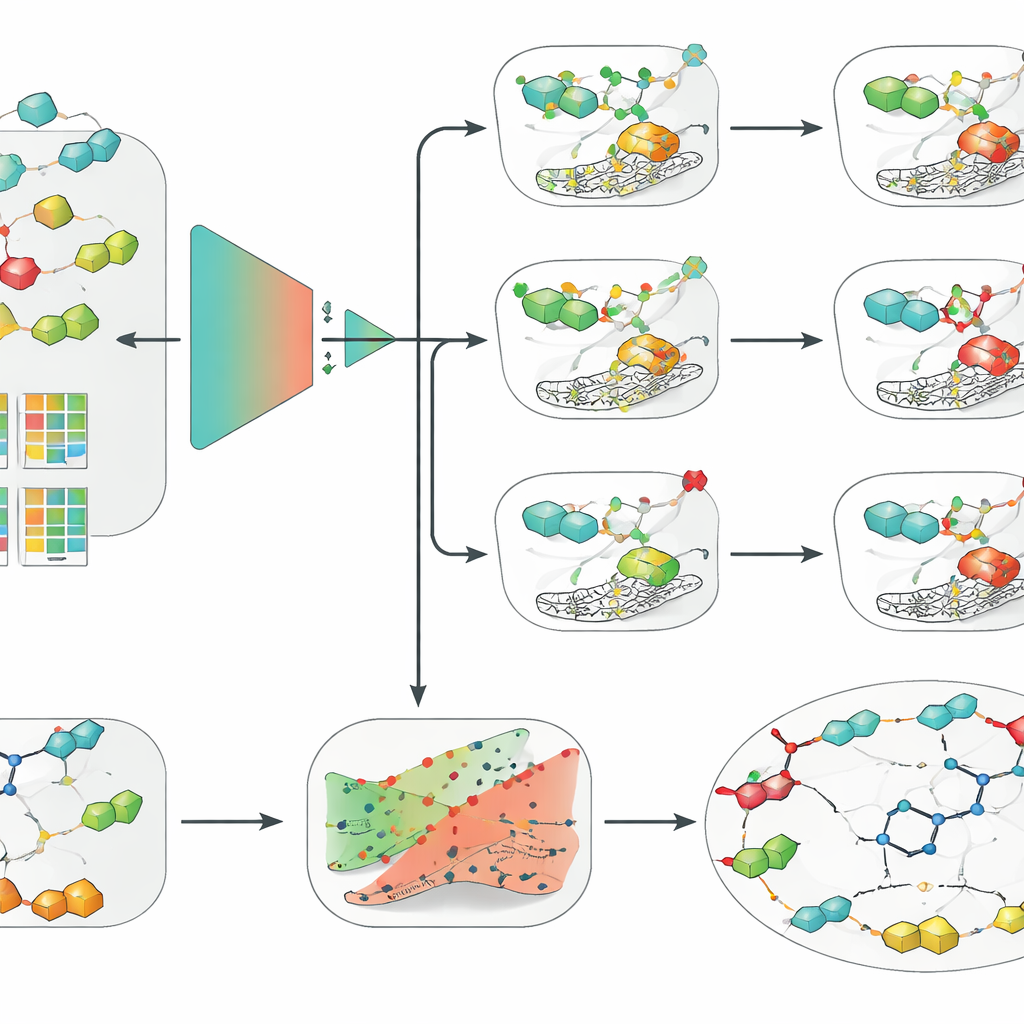
Författarna tacklade detta genom att först sammanställa en noggrant rensad databas med tusentals molekyler testade mot hCA II, IX och XII från ChEMBL-repositoriet. De standardiserade kemiska strukturer, tog bort tvivelaktiga mätningar och fokuserade på föreningar som delar en vanlig zinkbindande grupp typisk för denna klass av hämmare. Med strikta trösklar märkta de molekyler som tydligt aktiva eller tydligt inaktiva och sorterade bort gränsfall som skulle kunna förvirra modellerna. Eftersom det fanns betydligt fler inaktiva än aktiva molekyler balanserade de datan så att inlärningsalgoritmerna inte bara favoriserade majoritetsklassen. De använde också en "scaffold-baserad" uppdelning av datan så att tränings- och testset innehöll olika kärnramverk för molekyler, vilket ger en mer realistisk bild av hur väl modellerna hanterar verkligen nya föreningar.
Enkla modeller slår djupinlärning när data är begränsad
Med denna kurerade dataset jämförde teamet ett brett spektrum av metoder, från klassiska maskininlärningsmetoder som logistisk regression, random forests och supportvektormaskiner (SVM) till moderna djupa neurala nätverk, inklusive grafbaserade modeller som arbetar direkt på molekylstrukturer. De kombinerade dessa med flera sätt att koda molekyler, såsom traditionella handgjorda deskriptorer, nyckelbaserade fingeravtryck och inlärda inbäddningar från en kemisk språkmodell. Över alla tre enzymisoformer och under den striktare scaffold-baserade utvärderingen framstod en kombination konsekvent: en SVM matad med extended-connectivity fingerprints, ett strukturerat sätt att beskriva lokala kemiska omgivningar inom en molekyl. Förvånande nog överträffade denna relativt enkla uppsättning mer eleganta graf- och djupinlärningsmodeller, vilket betonar att datakvalitet, noggrann validering och bra molekylära deskriptorer kan vara viktigare än algoritmisk komplexitet när dataset är av måttlig storlek.
Lägga till pålitlig konfidens och människovänliga förklaringar
Forskarna förpackade sedan sin bästa SVM-modell i två ytterligare lager utformade för att göra dess prediktioner mer användbara i verklig läkemedelsupptäckt. Först tillämpade de ett ramverk kallat konformal prediktion, som inte bara ger ett enkelt ja-eller-nej-svar utan istället tillhandahåller ett intervall av sannolika utfall tillsammans med en garanterad felmarginal. Detta gör det möjligt för forskare att ställa in hur försiktig modellen ska vara och att känna igen fall där modellen är genuint osäker. För det andra använde de kontrafaktiska förklaringar för att göra modellens resonemang mer intuitivt. För en given molekyl genererade de nära besläktade analoga föreningar som vänder det förutsagda utfallet från aktivt till inaktivt eller vice versa. Genom att granska dessa par för den kliniska kandidaten SLC-0111, som selektivt blockerar IX och XII men inte II, återupptäckte metoden oberoende en viktig insikt från medicinsk kemi: små förändringar i molekylens "svans" påverkar starkt vilken isoform den föredrar att binda.
Från algoritmer till praktiska verktyg för läkemedelsdesign
För att göra sitt tillvägagångssätt tillgängligt paketade författarna de tre SVM-modellerna, osäkerhetslagret och den kontrafaktiska motorn i ett grafiskt verktyg kallat CAInsight. En användare kan ange en molekyls textrepresentation och med ett enda klick få förutsagd aktivitet mot hCA II, IX och XII, en uppskattning av hur pålitlig varje prediktion är och föreslagna strukturella justeringar som kan öka eller minska aktiviteten. Medan modellerna fokuserar på att klassificera molekyler som aktiva eller inaktiva snarare än att i ett steg förutsäga exakt potens eller selektivitet, reproducerar de redan känt beteende för verkliga läkemedelskandidater och särskiljer subtila strukturella förändringar. Författarna noterar att större och mer enhetliga dataset, plus en djupare analys av hur aktivitetsgränser väljs, skulle kunna förfina prestandan ytterligare.
Vad detta betyder för framtida cancerläkemedel
På ett enkelt sätt visar detta arbete att noggrant byggda och väl förklarade maskininlärningsmodeller kan hjälpa kemister att designa cancerläkemedel som bättre skiljer mellan likartade enzymmål. Genom att kombinera robust statistik, osäkerhetsuppskattningar och intuitiva "what-if"-exempel förutsäger ramverket inte bara vilka molekyler som sannolikt fungerar utan antyder också varför. Denna typ av transparent artificiell intelligens kan snabba upp virtuell screening, stödja generativ design av nya föreningar och minska prov-och-fel-bördor i laboratoriet, vilket i slutändan kan underlätta upptäckten av mer selektiva och säkrare behandlingar för patienter.
Citering: Ghamsary, M.S., Rayka, M. & Naghavi, S.S. Interpretable machine learning rationalizes carbonic anhydrase inhibition via conformal and counterfactual prediction. Sci Rep 16, 8419 (2026). https://doi.org/10.1038/s41598-026-39771-2
Nyckelord: hämmare av karbonanhydras, tolkbar maskininlärning, läkemedelsselektivitet, konformal prediktion, kontrafaktiska förklaringar