Clear Sky Science · sv
Komplexa heterozygota CHAT-genmutationer, en missense- och en splice-site-variant, hos två syskon med medfödd myasteniskt syndrom
När andningen plötsligt sviktar
Vissa barn verkar friska vid födseln men slutar plötsligt att andas vid lindriga feberanfall och behöver akut ventilationshjälp. För deras familjer är episoderna skrämmande och oförklarliga. Denna studie undersöker två sådana syskon från Japan och spårar deras livshotande svaghets- och apnéanfall (andningsuppehåll) till små förändringar i en enda gen som hjälper nerver att kommunicera med muskler. Genom att pussla ihop kliniska ledtrådar, gen-sekvensering och datorbaserad proteinmodellering visar forskarna hur dessa mutationer sannolikt stör ett nyckelenzym och ger läkare ett tydligare mål för diagnos och behandling.
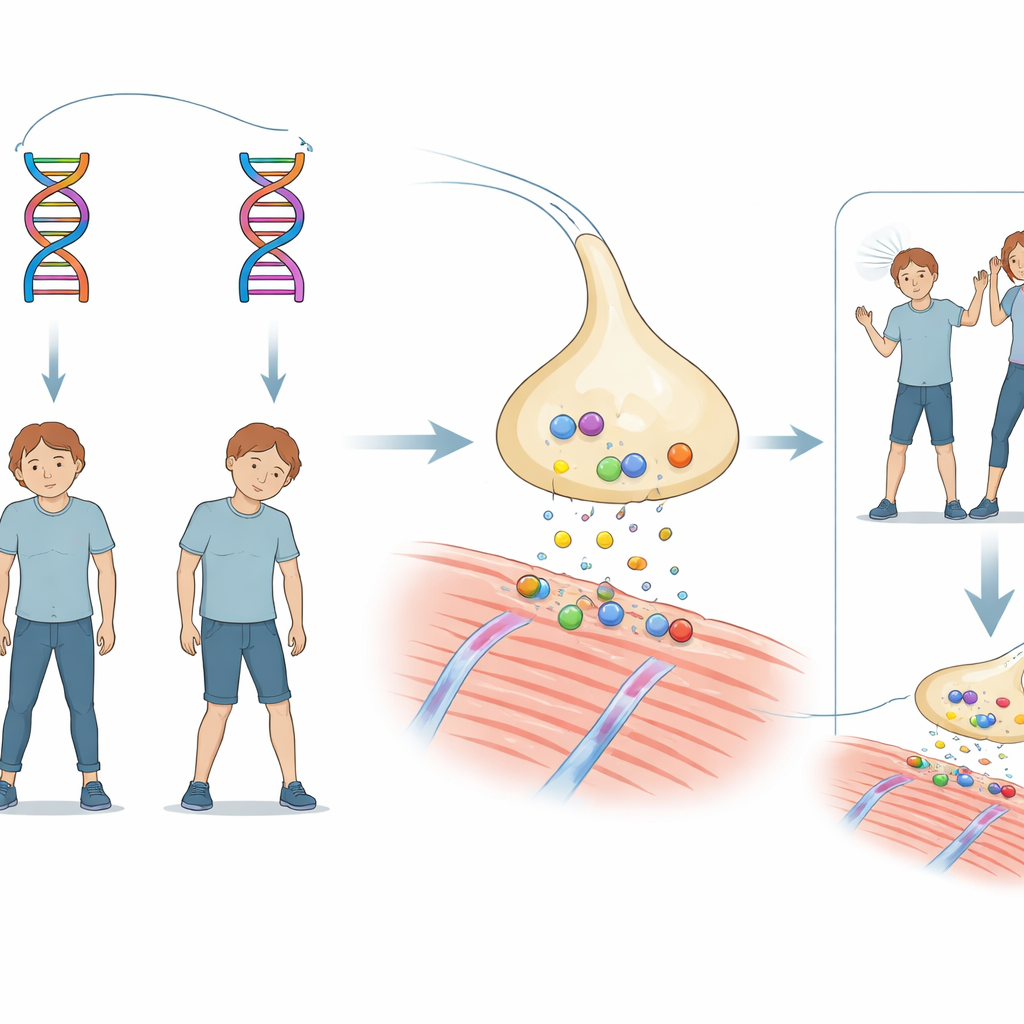
En familjegåta om plötslig svaghet
Berättelsen kretsar kring en bror och syster som båda hade något försenad motorisk utveckling i spädbarnsåldern. Runt 18 månaders ålder upplevde båda episoder med apné och medvetslöshet i samband med feber, så allvarliga att respirator blev nödvändig. När de växte fortsatte båda att drabbas av hängande ögonlock och generell muskelsvaghet utlösta av infektioner, feber eller ansträngning. Hjärnavbildningar var normala och vanliga antikroppsdrivna former av myasteni (en sjukdom där signaleringen mellan nerv och muskel är störd) uteslöts. En läkemedelsbehandling som ökar den kemiska signalen mellan nerver och muskler förbättrade dock tydligt deras symtom, vilket pekade mot ett sällsynt ärftligt tillstånd kallat medfödd myasteniskt syndrom.
Att hitta de felaktiga instruktionerna
För att leta efter en ärftlig orsak sekvenserade teamet alla protein‑kodande gener hos syskonen och deras föräldrar. De fann att varje barn bar på två olika förändringar i samma gen, CHAT, som kodar för kolinacetyltransferas—ett enzym som tillverkar acetylkolin, den huvudkemiska budbäraren som nerver använder för att aktivera muskler. En förändring bytte ut en enda byggsten i enzymet (en missense-mutation känd som G411R). Den andra låg vid en kritisk gräns där cellen normalt klipper och sammanfogar gensegment vid RNA‑processning (en splice-site-mutation betecknad c.752+2T>C). Varje förälder bar bara en av dessa förändringar och var frisk; endast barnen som ärvt båda visade sjukdom, vilket tyder på att kombinationen av mutationer tillsammans försvagar enzymets funktion.
Undersöka hur ett dolt klipp förändrar enzymet
Eftersom forskarna inte kunde få tillräckligt med naturligt CHAT‑RNA från blodceller använde de ett ”minigen”-experiment. De klonade den relevanta delen av genen i en DNA-vektor, införde antingen den normala eller den muterade versionen i odlade celler och undersökte sedan hur RNA bearbetades. I den normala konstruktionen innehöll RNA alla förväntade segment. I den muterade versionen hoppades ett helt segment, känt som exon 5, över, men läsramen för genen förblev intakt. Det betyder att enzymet skulle tillverkas men sakna ett kort internt stycke aminosyror. Jämförelser över arter visade att denna saknade region är starkt bevarad, vilket antyder att den har en viktig strukturell funktion.
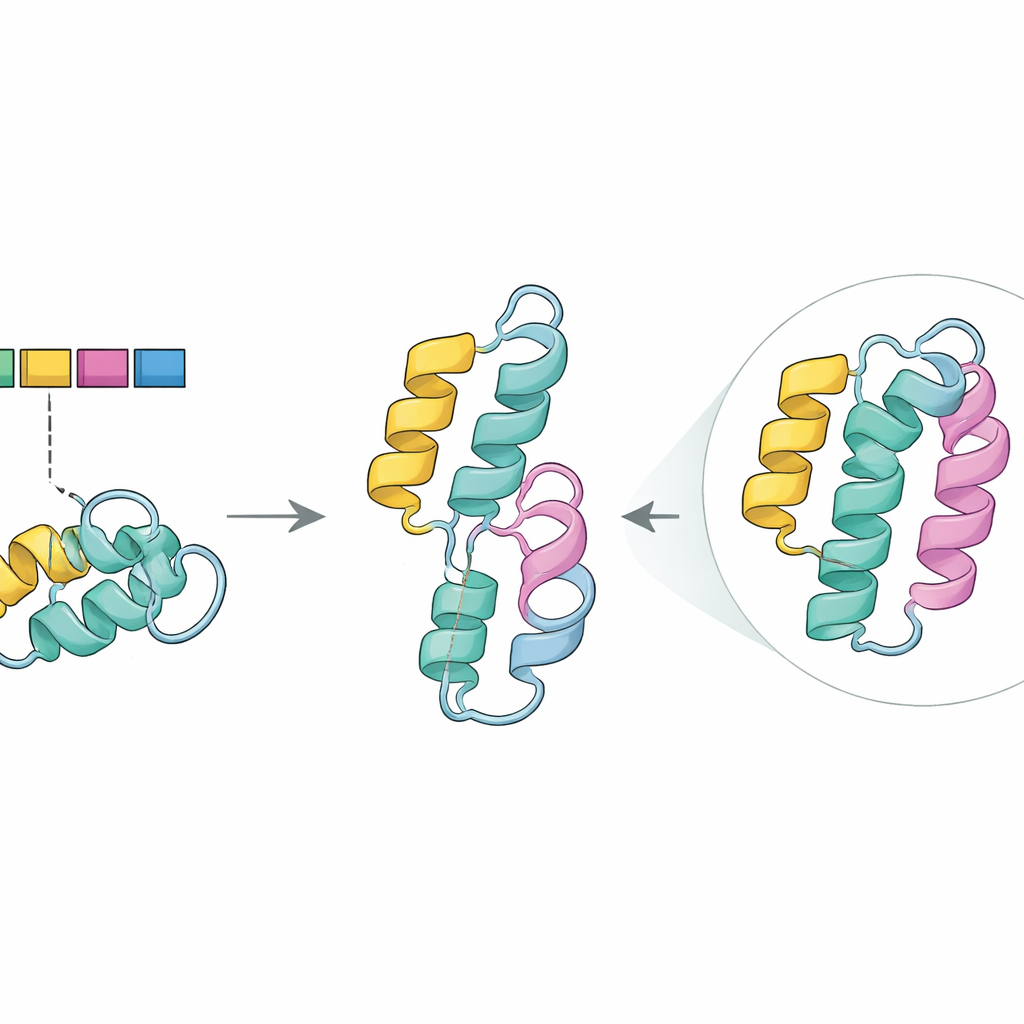
Se strukturskador in silico
För att utforska den funktionen vände sig teamet till AlphaFold2, ett avancerat program som förutspår tredimensionella proteiner från deras sekvenser. I det normala enzymet bildar delen som kodas av exon 5 en av de tätt packade spiralsegmenten (en alfa-helix) som hjälper till att stabilisera proteinets kärna. I den förutsagda mutanta strukturen försvann denna helix och lämnade ett gap i ett område som tidigare arbete visat är avgörande för att upprätthålla stabilitet och stödja effektiv kemi. Tillsammans med datorverktyg som pekar ut skadliga mutationer stöder dessa resultat idén att hoppet över exon 5, särskilt i kombination med G411R‑förändringen på den andra genkopian, försvagar enzymets prestanda utan att helt eliminera den—i linje med syskonens måttliga men allvarliga symtom.
Vad detta innebär för patienter och familjer
Studien sluter att kombinationen av G411R‑missensemutationen och den nyidentifierade splice-site‑mutationen i CHAT mycket sannolikt är ansvarig för syskonens medfödda myasteniska syndrom. Genom att visa, via minigen‑assayet och strukturell modellering, hur splice-site‑ändringen avlägsnar en stabiliserande helix från enzymet, ger författarna en mekanistisk förklaring som kliniker och forskare kan bygga vidare på. För drabbade familjer erbjuder sådant arbete mer än en diagnos: det stödjer skräddarsydd behandling med läkemedel som ökar neuromuskulär signalering, vägleder genetisk rådgivning inför framtida graviditeter och lägger till ett viktigt nytt exempel i katalogen över hur subtila förändringar i vår genetiska kod kan påverka muskelfunktion och den grundläggande handlingen att andas.
Citering: Kikuchi, S., Wada, N., Mariya, T. et al. Compound heterozygous CHAT gene mutations, a missense and a splice site variant, in two siblings with congenital myasthenic syndrome. Sci Rep 16, 9346 (2026). https://doi.org/10.1038/s41598-026-39759-y
Nyckelord: medfödd myasteniskt syndrom, CHAT-gen, kolinacetyltransferas, splice-site-mutation, neuromuskulär synaps