Clear Sky Science · sv
Matematisk modellering och beräkning av NM‑polynomindex för förutsägelse av fysikalisk‑kemiska egenskaper
Varför detta är viktigt för framtida läkemedel
Att utforma ett nytt läkemedel liknar till viss del att designa ett flygplan: du vill veta hur det kommer att bete sig långt innan du bygger det i verkligheten. För läkemedel omfattar detta hur lätt de avdunstar, hur de blandar sig med vatten eller fett och hur de rör sig genom kroppen. Denna artikel visar hur noggrant utformad matematik kan förutsäga många av dessa fysikalisk‑kemiska egenskaper enbart utifrån ett läkemedels struktur, vilket potentiellt sparar tid, kostnader och trial‑and‑error i läkemedelsupptäckt. 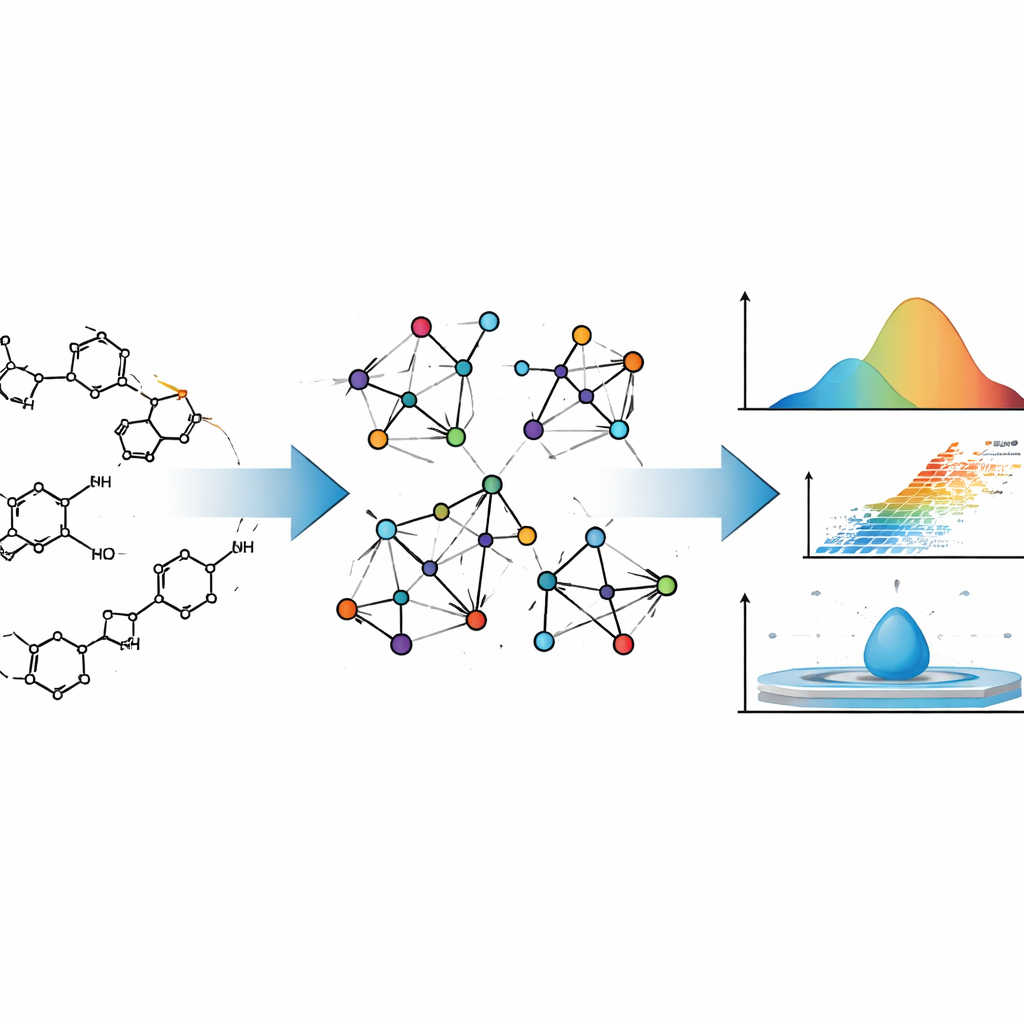
Från molekyler till nätverk
Författarna behandlar läkemedelsmolekyler inte bara som samlingar av atomer, utan som nätverk. I denna bild är varje atom en punkt och varje kemisk bindning en linje som förbinder två punkter. Denna typ av beskrivning kommer från grafteori, en gren av matematiken som studerar nätverk av alla slag, från länkar i sociala medier till elnät. Kemister har använt sådana "molekylära grafer" i årtionden, eftersom vissa numeriska sammanfattningar av dessa grafer — kallade topologiska index — ofta speglar hur molekyler beter sig i verkligheten, till exempel hur lätt de kokar eller hur täta de är.
Lägga till detalj om omgivningen
Traditionella index beaktar vanligtvis bara hur många bindningar som vidrör varje atom. Teamet bakom denna studie går ett steg längre. De använder så kallade neighborhood M‑polynomial (NM‑polynomial) index, som inte bara räknar en atoms egna kopplingar utan också sammanfattar hur anslutna dess grannar är. Denna rikare beskrivning fångar nyanser som hur förgrenad en molekyl är, hur dess ringar är sammanfogade och var syre‑ eller kväveatomer befinner sig i strukturen. Dessa egenskaper påverkar i sin tur hur starkt molekyler binder till varandra, hur styva de är och hur deras elektroner reagerar på elektriska fält — alla viktiga ingredienser i fysikalisk‑kemiska egenskaper.
Testa idén på verkliga cancerläkemedel
För att förankra sin matematik i verkligheten räknar författarna först ut NM‑polynomindex för två välkända anticancerläkemedel, Mitoxantron och Doxorubicin. Båda är komplexa, fler-ringade molekyler som används i stor utsträckning i kemoterapi. Genom att översätta deras detaljerade kemiska ritningar till molekylgrafer och vidare till NM‑polynomindex visar författarna hur metoden systematiskt följer strukturella förändringar över olika "storlekar" av dessa molekyler. De automatiserar sedan processen med ett Python‑program som tar en molekyls kopplingsinformation (i form av en adjacensmatris) och omedelbart returnerar hela uppsättningen index, vilket minimerar mänskliga fel och påskyndar beräkningar som skulle vara mödosamma för hand. 
Träna maskiner att läsa molekylära fingeravtryck
Nästa steg går bortom dessa två läkemedel till en bredare samling av 45 polycykliska läkemedel, inklusive välkända namn som paracetamol, ibuprofen och flera moderna riktade terapier. För varje läkemedel sammanställer de nio NM‑polynomindex och nio experimentellt uppmätta egenskaper: komplexitet, kokpunkt, förångningsentalpi, antändningspunkt, molar refraktivitet, polariserbarhet, ytspänning, molarvolym och brytningsindex. De tränar sedan flera regressionsmodeller i maskininlärningsstil — Linear, Ridge, Lasso och Elastic Net — för att lära sig hur kombinationer av index kartlägger till varje egenskap. Noggranna statistiska säkerhetsåtgärder används genomgående: de tar bort redundanta indata, standardiserar variabler, utför upprepad korsvalidering på 80 % av datan och testar slutligen modellerna på en orörd 20 %.
Vad siffrorna visar
Modellerna visar att NM‑polynomindex är särskilt kraftfulla för egenskaper som hänger ihop med hur molekyler packas och interagerar. För kokpunkt, förångningsentalpi, antändningspunkt, molar refraktivitet, polariserbarhet och molarvolym når de bästa modellerna mycket höga korrelationspoäng, vilket innebär att de förutsagda värdena följer de experimentella tätt. Regulariserade metoder som Ridge och Elastic Net presterar generellt bäst, vilket tyder på att en mild inskränkning hjälper modellerna att fokusera på de mest informativa aspekterna av indexen. En korrelations‑heatmap bekräftar att flera index — särskilt de relaterade till övergripande konnektivitet och "närskapsrikedom" — är starkt och konsekvent kopplade till dessa egenskaper över panelen med 45 läkemedel.
Begränsningar och möjligheter till förbättring
Inte alla egenskaper samarbetar. Brytningsindexet, som fångar hur ljus böjs när det går in i ett material, visar sig vara besvärligt: modellerna har svårt att göra bättre än enkla medelvärden och NM‑polynomindexen uppvisar bara svaga korrelationer med det. Ytspänningen fångas måttligt bra, men inte lika tydligt som de andra egenskaperna. Dessa luckor antyder att vissa beteenden beror på egenskaper bortom tvådimensionell konnektivitet, såsom tredimensionell form eller subtila elektroniska effekter. Författarna föreslår att framtida arbete kan slå ihop NM‑polynomindex med kvantkemiska eller 3D‑deskriptorer för att överbrygga detta gap.
Vad detta betyder för läkemedelsdesign
Kort sagt visar studien att sofistikerad men välstrukturerad matematik kan omvandla en statisk skiss av en molekyl till en förvånansvärt exakt förutsägare av hur den molekylen beter sig i labbet. För många viktiga egenskaper — hur svårt det är att få den att koka, hur skrymmande den är eller hur lätt dess elektroner förskjuts — matchar eller överträffar NM‑polynommetoden, i kombination med moderna regressionsmetoder, tidigare metoder som använde enklare index eller mindre datamängder. Även om den ännu inte ersätter experiment helt, erbjuder den läkemedelsdesigners ett snabbare siktverktyg: genom att beräkna dessa grafbaserade fingeravtryck kan de tidigt uppskatta viktiga fysikalisk‑kemiska egenskaper, rikta laboratoriearbetet mot de mest lovande kandidaterna och utforska kemisk rymd mer effektivt.
Citering: Tawhari, Q.M., Naeem, M., Koam, A.N.A. et al. Mathematical Modeling and Computation of NM-Polynomial Indices for Physicochemical Properties Prediction. Sci Rep 16, 8136 (2026). https://doi.org/10.1038/s41598-026-39562-9
Nyckelord: kemisk grafteori, prediktion av läkemedelsegenskaper, molekylär topologi, maskininlärning i kemi, fysikalisk‑kemiska deskriptorer