Clear Sky Science · sv
Beräkningsoptimering av löslighet i DEK1 calpain-domän genom integrerad strukturell modellering och datadriven riktad mutagenes
Varför det spelar roll att få växtproteiner att uppföra sig
Många av de proteiner som styr hur växter växer är stora, känsliga molekyler som vägrar lösa sig när forskare försöker studera dem i laboratoriet. Ett sådant protein, kallat DEK1, hjälper till att forma växtens kropp från cellnivå uppåt. Men eftersom en avgörande del av DEK1 klumpar ihop sig när den produceras i bakterier har dess tredimensionella struktur förblivit okänd, vilket bromsar ansträngningar att förstå och utnyttja den. Denna studie visar hur datorbaserad modellering och smart, datadriven design kan omforma den problematiska regionen så att den blir mer löslig utan att förstöra dess uppbyggnad — och erbjuder en generell metod för att tygla svårhanterliga proteiner.
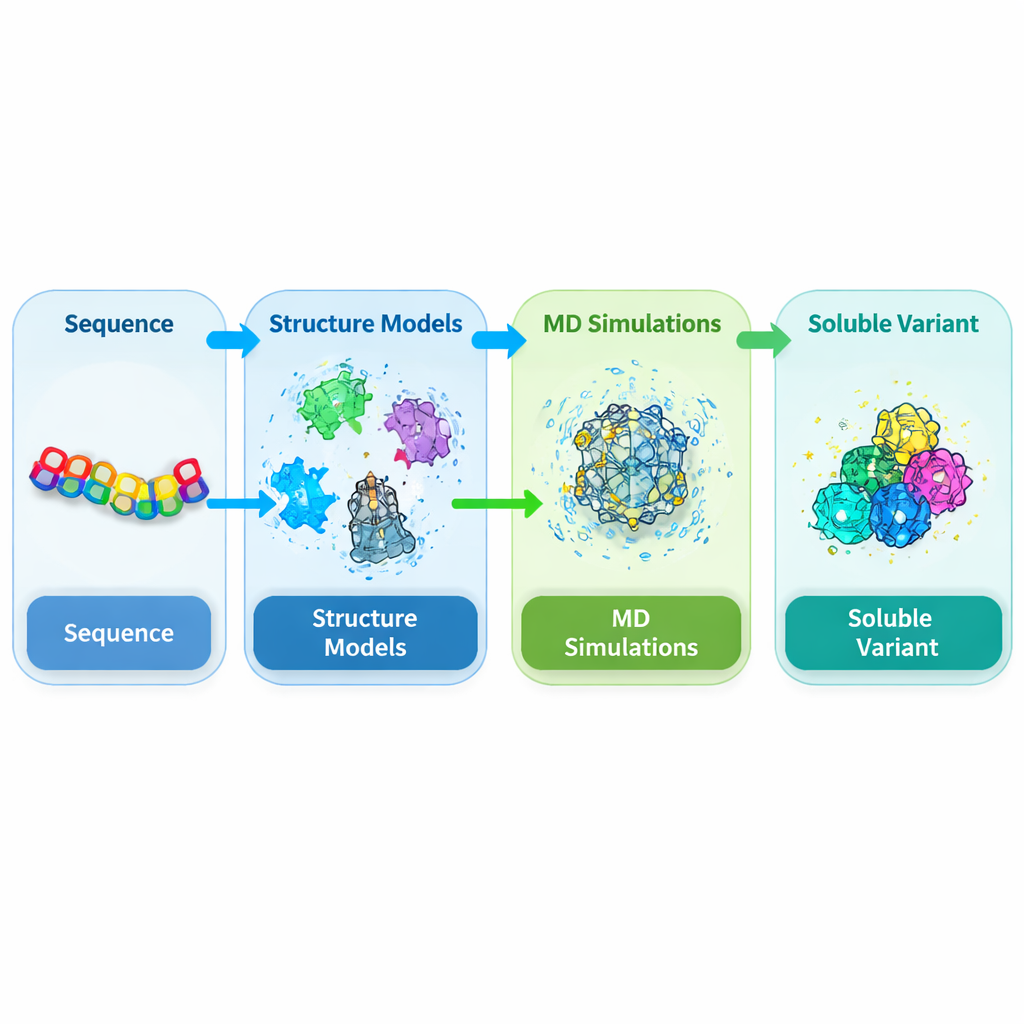
Att rikta in sig på problemområdet i ett nyckelprotein hos växter
DEK1 är ett ovanligt stort protein inbäddat i cellmembran och avslutat av en klyvande enzymregion känd som en calpain-domän. Genetiska studier har visat att denna domän är nödvändig för normal utveckling i växter såsom mossor och grödor, men dess struktur har aldrig lösts experimentellt. När forskare försöker framställa denna calpain-kärna (kallad CysPc) i den vanliga värdbakterien Escherichia coli tenderar den att bli olöslig och bilda täta inklusionskroppar. Det gör det nästintill omöjligt att rena i de mängder och den kvalitet som krävs för detaljerade strukturella och funktionella studier. Författarna bestämde sig därför för att omdesigna CysPc-domänen så att den skulle lösa sig lättare samtidigt som dess övergripande form bevarades.
Bygga en pålitlig 3D-modell från grunden
Eftersom det inte finns någon experimentell struktur för denna växtcalpain var teamet först tvunget att förutsäga dess tredimensionella form. De kombinerade flera toppmoderna strukturförutsägelseverktyg, inklusive AlphaFold2, SWISS-MODEL och I-TASSER, och förankrade dessa förutsägelser i kända strukturer från besläktade kalpainer hos däggdjur. Genom en konsensusmetod förfinade och kontrollerade de resulterande modellerna med flera kvalitetskontroller som bedömer ryggradens geometri, packning och överensstämmelse med kända strukturella mönster. Dessa oberoende kontroller visade att den integrerade modellen av CysPc-domänen var mer tillförlitlig än någon enskild förutsägelse ensam, vilket gav en stabil utgångspunkt för att utforska hur små förändringar i aminosyrasekvensen kunde förbättra lösligheten.
Testa virtuella mutationer i ett simulerat lösningsmedel
Med 3D-modellen i handen körde författarna omfattande molekylärdynamiksimuleringar där proteinet och de omgivande vattenmolekylerna följdes över tid på datorn. De fokuserade på rester på proteinets yta som var flexibla, hydrofoba eller förutspådda att främja aggregering. Kandidatpositioner muterades individuellt till mer vattenvänliga aminosyror och simulerades sedan i 200 nanosekunder vardera. För varje variant mätte de egenskaper relaterade till löslighet, såsom hur stor yta som är i kontakt med vatten, hur kompakt proteinet förblev och hur starkt atomer fluktuerade. Många enstaka mutationer ökade måttligt solventexponering eller interna vätebindningar samtidigt som den övergripande formen förblev oförändrad, vilket tyder på att CysPc:s grundläggande stomme kunde tåla noggrant utvalda substitutioner.
Låta algoritmer söka i mutationsrymden
Att ändra bara en rest ger sällan dramatiska vinster i löslighet, så forskarna undersökte nästa kombinationer av två och tre mutationer. De skapade ett bibliotek av dubbel- och trippelvarianter byggda från de bästa enkelmutationerna och simulerade återigen varje variant. För att poängsätta och rangordna dessa designer rättvist definierade de ett viktat index som kombinerar flera simuleringsfunktioner kända för att korrelera med löslighet, som belönar ökad hydrering och intern bindning samtidigt som överdriven flexibilitet bestraffas. De använde sedan en förstärkningsinlärningsalgoritm (Proximal Policy Optimization) för att navigera i den enorma rymden av möjliga trippelmutanter och föreslå de mest lovande kombinationerna. Denna datadrivna sökning konvergerade mot en specifik trippelmutant, kallad MUT347, som toppkandidat.
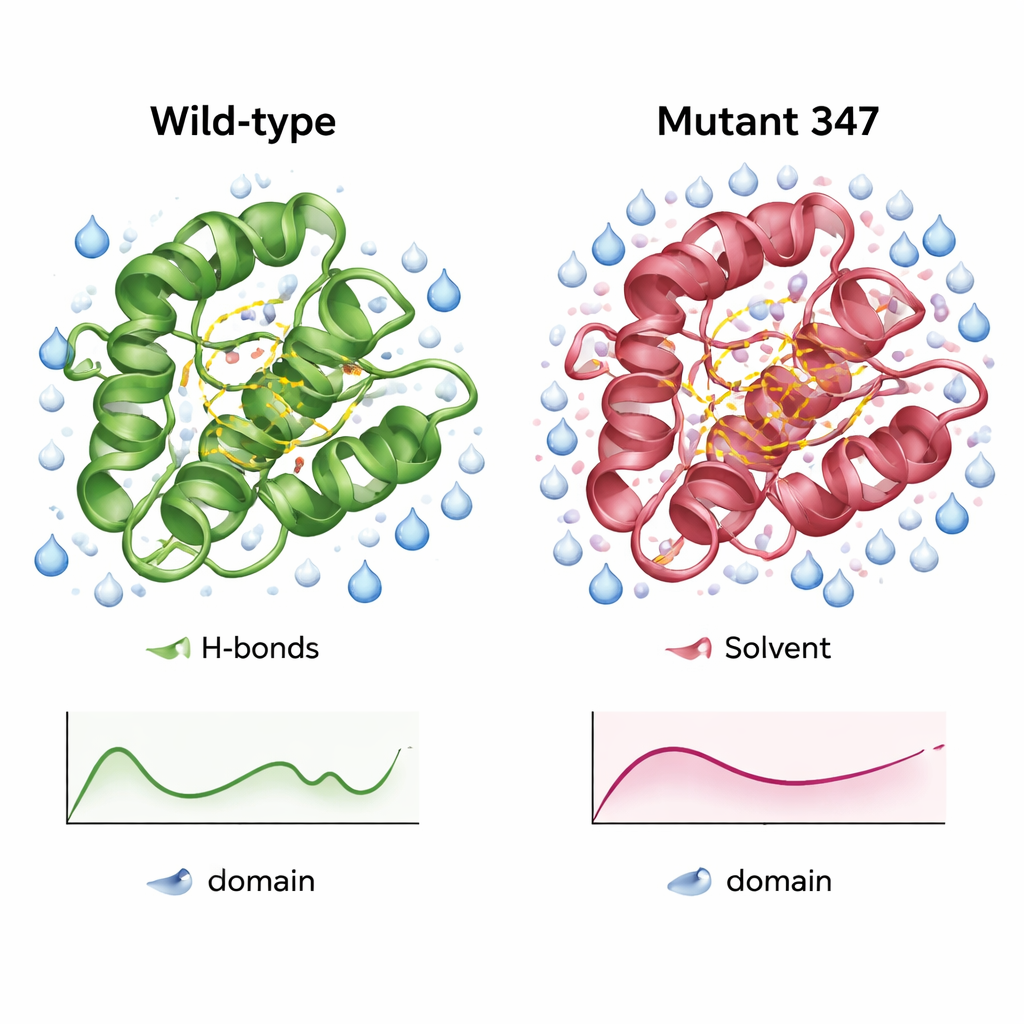
En mer kompakt, bättre hydrerad version av enzymet
Detaljerade simuleringar av vildtypens CysPc-domän och MUT347 visade hur den konstruerade varianten skiljde sig. MUT347 nådde jämvikt snabbare och visade mindre totala avvikelser från sin startform, vilket indikerar större strukturell stabilitet i lösning. Dess slingor och kedjeändar var något mindre tillgängliga, medan den katalytiska kärnan behöll sin ursprungliga rörlighet, vilket tyder på att funktionellt viktiga rörelser bevarades. Trippelmutanten hade fler interna vätebindningar och en större vattenåtkomlig yta i nyckelregioner, tecken på en bättre organiserad och mer hydrerad yta. Vid varierande salthalter och pH-nivåer upprätthöll MUT347 konsekvent lägre fluktuationer än det ursprungliga proteinet, ett beteende som förknippas med minskad benägenhet att klumpa ihop sig.
Vad detta betyder för att studera och återanvända proteiner
För icke-specialister är slutsatsen att författarna har byggt ett i stort sett datorbaserat recept för att förvandla en klumpig, klibbande del av ett viktigt växtprotein till en mer löslig, välfungerande version utan att först ha en experimentellt bestämd struktur. Genom att kombinera moderna strukturförutsägelser, långsiktiga simuleringar och inlärningsalgoritmer som kan hantera många designval samtidigt identifierade de en trippelmutation som förutsägs stabilisera veckningen och exponera den mer gynnsamt för vatten. Även om experimentella studier fortfarande behövs för att bekräfta vinsterna i verkliga provrör, kan detta ramverk vara brett användbart för att rädda andra eukaryota proteiner som är svåra att producera, och i förlängningen hjälpa forskare att låsa upp strukturer och funktioner som idag är utom räckhåll.
Citering: Dabiri, M., Levarski, Z., Struhárňanská, E. et al. Computational optimization of DEK1 calpain domain solubility through integrated structural modelling and data-driven targeted mutagenesis. Sci Rep 16, 7767 (2026). https://doi.org/10.1038/s41598-026-38805-z
Nyckelord: proteinslöslighet, beräkningsmutagenes, molekylär dynamik, växtcalpain DEK1, proteinteknik