Clear Sky Science · sv
LncRNA FTX främjar myokardfibros genom att svälja miR-335-3p för att reglera TFEC/ILK-signalering
Varför hjärtscarring spelar roll
Hjärtsvikt drabbar tiotals miljoner människor globalt och utvecklas ofta tyst över flera år. En huvudorsak till detta försämrade tillstånd är myokardfibros—långsam, progressiv ärrbildning i hjärtmuskeln som gör den stelare och mindre kapabel att pumpa blod. Denna studie undersöker det molekylära ”kopplingsschemat” som får hjärtceller att lägga ner för mycket ärrvävnad, och identifierar en ny molekylkedja som skulle kunna riktas för att bromsa eller till och med vända denna skadliga process.
Närmare titt på hjärtscarring
När hjärtat skadas eller utsätts för stress går stödjeceller kallade hjärtfibroblaster in i högvarv. Vid normal reparation hjälper de till att laga skador. Men vid kronisk sjukdom kan de växla till ett överaktivt tillstånd och producera överskott av kollagen och andra komponenter i extracellulärmatrixen, vilket i längden gör hjärtväggen stel. Forskarna använde två modeller för att studera denna process: möss behandlade med läkemedlet isoprenalin, som pålitligt inducerar hjärtfibros, och mänskliga hjärtfibroblaster exponerade för signalmolekylen TGF-β1, en välkänd trigger för ärrbildning. I båda systemen mätte de hur specifika gener och proteiner förändrades när fibros utvecklades.
En skadlig kedjereaktion inne i cellerna
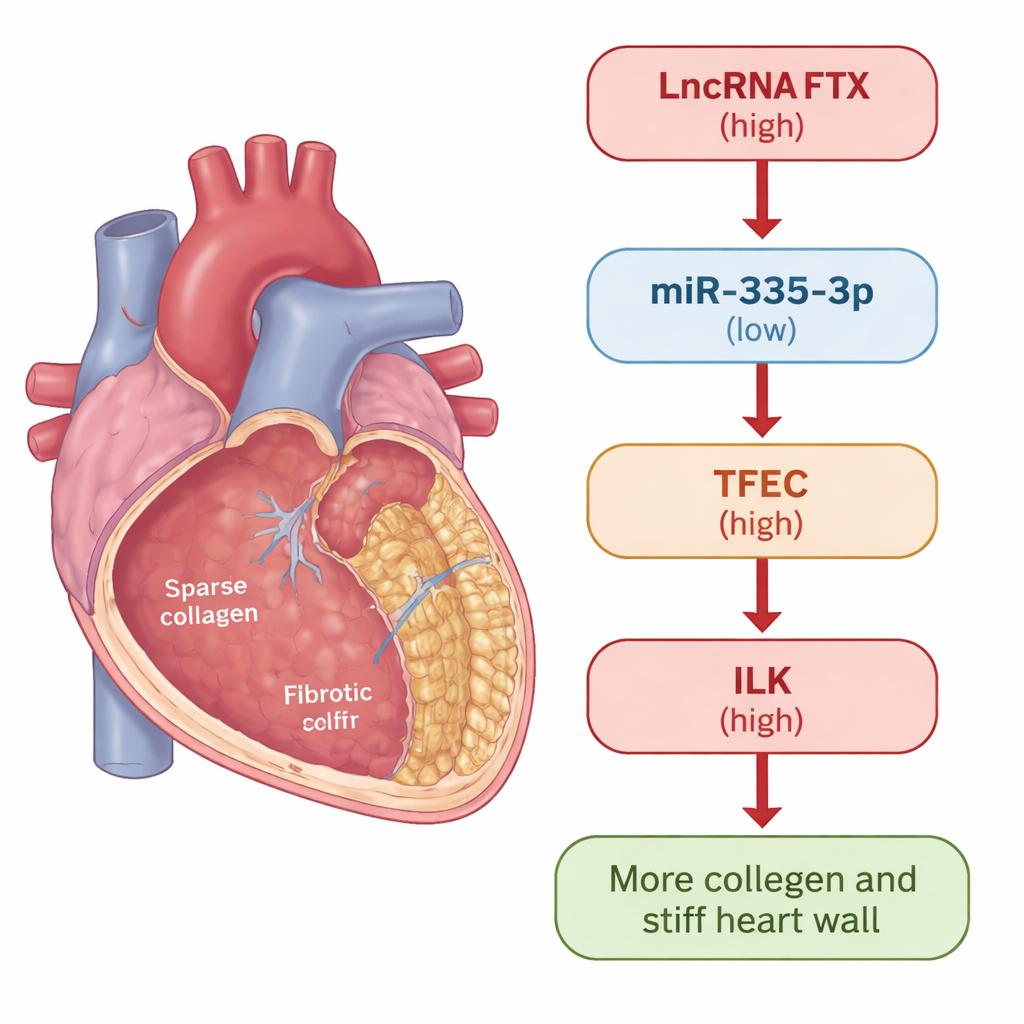
Teamet fokuserade på en transkriptionsfaktor kallad TFEC, ett protein som sitter i cellkärnan och aktiverar andra gener. De fann att TFEC, tillsammans med ett annat protein kallat integrin-linked kinase (ILK), konsekvent ökade när fibroblaster drevs mot ett fibrotiskt, ärrbildande tillstånd. Att tysta antingen TFEC eller ILK minskade kraftigt klassiska fibrosmarkörer såsom α-smooth muscle actin och kollagenerna I och III, och dämpade även en tillväxtkontrollväg (Akt/GSK3β och Hippo-signalering) känd för att främja vävnadsärr. Experiment som kartlade DNA-bindning visade att TFEC direkt binder till promotorn för ILK-genen och ökar dess aktivitet, vilket placerar TFEC tydligt uppströms om ILK i en pro-fibrotisk signalväg.
RNA-brytare som styr huvudregulatorn
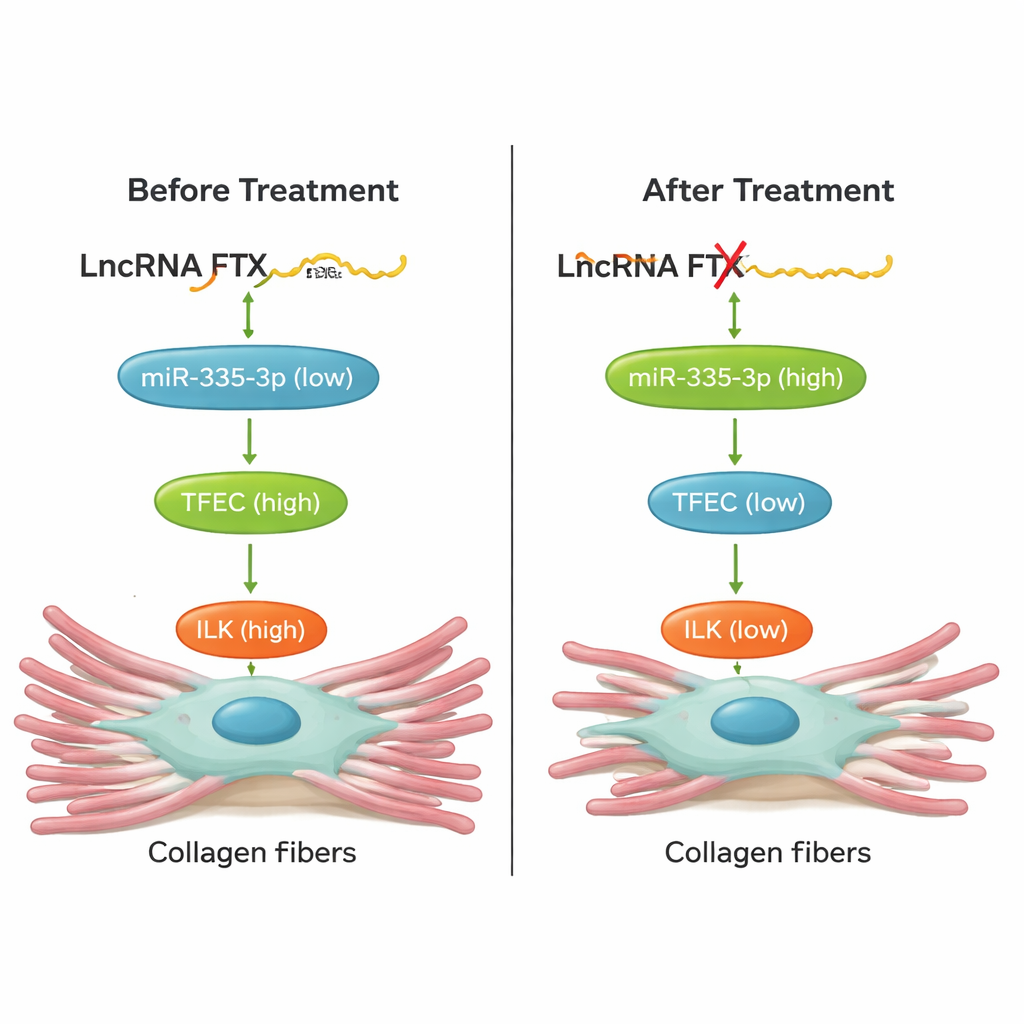
För att förstå vad som styr TFEC vände forskarna sig till icke-kodande RNA—RNA-molekyler som inte bildar proteiner men fungerar som finjusterare av genaktivitet. De identifierade ett litet RNA, miR‑335‑3p, som var minskat i fibrotiska hjärtan och celler. Att höja miR‑335‑3p-nivåerna sänkte TFEC, medan att blockera det ökade TFEC, och rapportörtester bekräftade att miR‑335‑3p binder direkt till TFEC-mRNA för att hålla dess nivåer i schack. De fann sedan ett långt icke-kodande RNA, kallat FTX, som var förhöjt vid fibros och fysiskt interagerade med miR‑335‑3p. FTX fungerade som en molekylär svamp: det sugade upp miR‑335‑3p och hindrade detta lilla RNA från att dämpa TFEC. Som en följd ökade TFEC och ILK, och fibroblasterna producerade mer ärrbildande kollagen.
Från cellodling till levande hjärtan
Avgörande testade teamet om störning av denna kedja faktiskt kunde skydda hjärtan hos djur. I möss som exponerades för isoprenalin ledde nedreglering av TFEC, knockdown av FTX i hjärtat med en AAV9-genterapivektor, eller förstärkning av miR‑335‑3p med en kemiskt stabiliserad ”agomir” alla till mindre kollagenuppbyggnad och lägre nivåer av fibrosmarkörer i hjärtvävnad. Dessa interventioner förbättrade också hjärtfunktionen: slagvolym och ejektionsfraktion rörde sig tillbaka mot normalvärden, och skadliga ökningar i hjärtfrekvensen dämpades. Räddningsexperiment i celler visade att ändring av en komponent i FTX/miR‑335‑3p/TFEC/ILK-axeln förutsägbart påverkade de andra, vilket bekräftar att detta är en tätt kopplad väg snarare än en lös korrelation.
Vad detta betyder för framtida behandlingar
För icke-specialisten är huvudbudskapet att författarna har identifierat en ny ”kontrollspak” för hjärtscarring. Ett långt RNA kallat FTX släpper bromsen (miR‑335‑3p) från en huvudomkopplare (TFEC), som sedan slår på ILK och nedströms pro-ärrsignalering, vilket driver överdriven kollagennedläggning och hjärtstelhet. Genom att minska FTX, återställa miR‑335‑3p eller blockerar TFEC direkt var det i möss möjligt att reducera ärrbildning och förbättra pumpfunktionen. Även om mer arbete krävs för att bekräfta denna väg hos mänskliga patienter och utveckla säkra terapier, erbjuder denna RNA-baserade regulatoriska kedja flera lovande mål för att ingripa i fibrosdriven hjärtsvikt.
Citering: Yao, F., He, Z., Zheng, C. et al. LncRNA FTX promotes myocardial fibrosis by sponging miR-335-3p to regulate TFEC/ILK signaling. Sci Rep 16, 7340 (2026). https://doi.org/10.1038/s41598-026-38615-3
Nyckelord: myokardfibros, hjärtsvikt, icke-kodande RNA, hjärtfibroblaster, fibros signalering