Clear Sky Science · sv
Felkarakterisering och felkorrigeringsmetoder i kombinatorisk DNA‑baserad lagring
Lagra världens data i DNA
Våra telefoner, servrar och molncenter drunknar i information, och traditionella lagringstekniker har svårt att hänga med. DNA — samma molekyl som bär genetisk information i levande organismer — erbjuder ett lockande alternativ: det är oerhört tätt packat, långlivat och kräver nästan ingen energi för att bevaras. Denna artikel undersöker en särskilt kraftfull variant av DNA‑datalagring, kallad kombinatorisk DNA‑kodning, och visar hur en ny typ av felkorrigering kan göra den mycket mer tillförlitlig i praktiken.
Hur man packar fler bitar i DNA
Konventionell DNA‑lagring skriver data genom att välja en av fyra baser (A, C, G, T) på varje position längs en syntetisk DNA‑sträng. Kombinatorisk DNA‑kodning tar ett annat grepp. Istället för att använda en enda kort DNA‑fragment på varje position, använder den omsorgsfullt utvalda kombinationer av korta fragment. Varje position i ett digitalt meddelande representeras inte av en sekvens utan av en uppsättning av dem hämtade ur ett fördefinierat bibliotek. Detta ökar kraftigt mängden information som kan packas in i varje syntessteg, vilket minskar kostnad och tid. Det innebär dock också att för att korrekt läsa en enskild ”bokstav” i det lagrade meddelandet måste systemet detektera alla fragment som borde vara närvarande på den positionen.
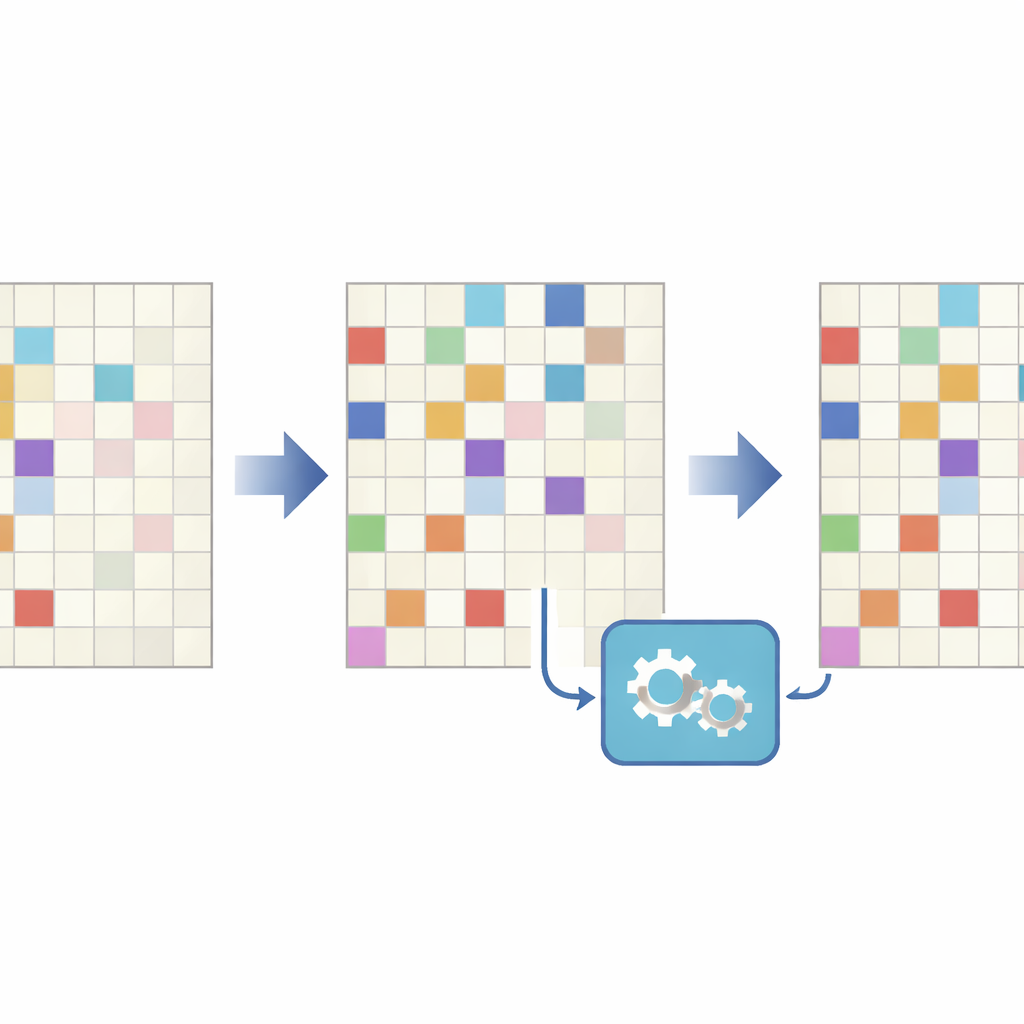
När vissa delar tyst försvinner
Eftersom DNA‑molekyler produceras och läses i stora antal uppträder samma kombinatoriska sekvens många gånger, varje kopia framställd och läst med små imperfektioner. Författarna granskade flera experimentella dataset och upptäckte att en särskild typ av fel dominerar i kombinatorisk DNA‑lagring: utplåning av ett enskilt fragment från en annars korrekt kombination. Med andra ord observeras helt enkelt inte en medlem av mängden i sekvenseringsavläsningarna, trots att de andra är närvarande. Dessa ”asymmetriska kombinatoriska utplåningar” blir särskilt vanliga när antalet avläsningar per lagrad sekvens är lågt — en realistisk situation i storskaliga system, där djupare sekvensering är kostsam. Under ungefär 50 avläsningar per sekvens ökar frekvensen av sådana saknade bitar snabbt, vilket gör det svårt eller omöjligt att rekonstruera de avsedda uppgifterna med standardmetoder.
Undersöka fel i större skala
För att gå bortom små demonstrationen samarbetade teamet med en industriell partner för att bygga ett stort proof‑of‑concept‑lagringssystem med kombinatorisk DNA. De kodade tusentals bitar text i 640 distinkta kombinatoriska sekvenser, var och en med åtta positioner som bär information. Specialiserade laboratorieprotokoll satte ihop pooler av DNA‑molekyler där varje molekyl representerade en kombination av korta fragment. Forskarna sekvenserade sedan miljontals avläsningar och använde en anpassad analyskedja baserad på BLAST, ett välkänt verktyg för sekvensjustering, för att identifiera vilka fragment som dök upp på varje position. Denna stora dataset bekräftade den tidigare observationen: när lästäckningen var hög kunde de flesta kombinationer rekonstrueras, men när det genomsnittliga antalet avläsningar per sekvens sjönk blev saknade fragment — och därmed utplåningsfel — det största hindret för korrekt avkodning.
En kod som förväntar sig envägsfel
Traditionella felkorrigeringsscheman som används i DNA‑lagring antar ofta att fel är ungefär symmetriska — symboler kan förväxlas, läggas till eller förloras med liknande sannolikhet. Den antagandet passar inte kombinatorisk DNA, där det typiska felet är att ett fragment som finns i originalkombinationen inte visar sig alls, medan störande extra fragment är relativt sällsynta. För att ta itu med detta utformade författarna en ny felkorrigeringskod, kallad en kombinatorisk VT‑kod, som är anpassad till detta envägsbeteende. De representerar varje kombinatoriskt tecken som en rad i en binär matris och behandlar saknade fragment som bitar som endast kan flippa från ett till noll. Koden använder ett matematiskt fingeravtryck, eller ”syndrom”, för varje tecken som kan avslöja vilket fragment som saknades, även när endast en del av kombinationen observeras. Dessa syndrom skyddas i sin tur av en Reed–Solomon‑kod, vilket möjliggör återställning av flera sådana fel över en sekvens.
Sätta den nya metoden på prov
Forskarna ställde sin skräddarsydda kod mot ett mer konventionellt tvådimensionellt Reed–Solomon‑schema som tidigare använts i DNA‑lagring. De testade båda i mjukvarusimuleringar och i ett andra storskaligt experiment, där hälften av sekvenserna skyddades av den traditionella metoden och hälften av den nya kombinatoriska koden, med identisk redundans. Över ett spektrum av förhållanden dominerade av utplåningsfel rekonstruerade den nya metoden oftare originaldata korrekt, och den presterade särskilt väl när lästäckningen var låg. Under dessa hårdare förhållanden misslyckades den traditionella metoden ofta med att avkoda hela sekvenser, medan den kombinatoriska VT‑schemat ändå återställde dem.
Varför detta spelar roll för framtida DNA‑arkiv
Arbetet visar att göra DNA‑datalagring praktisk inte bara handlar om att pressa in fler bitar i molekyler — det kräver också felkorrigering som matchar de verkliga felmönstren i de laboratorieprocesser som används. Genom att noggrant studera hur kombinatorisk DNA‑lagring fallerar, och genom att designa koder som specifikt förväntar sig att fragment saknas, demonstrerar författarna en tydlig väg mot mer tillförlitliga och skalbara DNA‑arkiv. När DNA‑baserade system växer för att hantera allt större datamängder kommer sådana skräddarsydda, asymmetriska felkorrigeringsstrategier att vara avgörande för att förvandla sköra molekylblandningar till pålitliga långtidsminnen.
Citering: Preuss, I., Sabary, O., Gabrys, R. et al. Error characterization and error correction approaches in combinatorial DNA-based storage. Sci Rep 16, 8093 (2026). https://doi.org/10.1038/s41598-026-38599-0
Nyckelord: DNA‑datalagring, felkorrigering, kombinatorisk kodning, utplåningsfel, informationsdensitet