Clear Sky Science · sv
Integrerade metoder för att utforska tidsmässiga och rumsliga förändringar i genåterblandning hos högpatogena fågelinfluensa A(H5)-virus i Eurasien, 2000–2023
Varför fågelinfluensas genetiska ”remixzoner” har betydelse för oss
Fågelinfluensa är inte längre bara ett problem för höns och ankor på avlägsna gårdar. En mycket farlig form av fågelinfluensa, känd som H5, har spridit sig över Europa och Asien i mer än två decennier, dödat vildfåglar, utplånat fjäderfäbesättningar och ibland infekterat däggdjur, inklusive kor och människor. Denna studie ställer en enkel men brådskande fråga: var och under vilka förhållanden är viruset mest benäget att ”remixa” sina gener och skapa nya, potentiellt farligare stammar — och hur kan vi upptäcka dessa farozoner i förväg?
Att följa ett formförändrande virus
Influensavirus bär sitt genetiska material i åtta separata delar, som kan bytas ut när två olika stammar infekterar samma fågel. Denna process, kallad reassortering, kan skapa helt nya viruskombinationer. Forskarna samlade in mer än 300 000 influensagensekvenser från globala databaser och, med hjälp av en standardiserad analyskedja, grupperade dem i genetiska familjer för varje av de åtta segmenten. De definierade sedan 136 distinkta genetiska ”genotyper” av högpatogena H5-virus som cirkulerade världen över mellan 1996 och 2023. Genom att spåra var och när dessa genotyper dök upp kunde de rekonstruera det föränderliga landskapet för H5-virus över tid.
Tre vågor av virusförändring
Teamet fann att H5:s evolution över Eurasien utvecklades i tre breda vågor. Från 2000 till 2013 dominerade en huvudgenotyp utbrotten, mestadels i Asien och delar av Afrika, vilket gav upphov till sporadiska men allvarliga händelser i fjäderfäbesättningar. Runt 2014 framträdde en ny gren av H5, känd som klad 2.3.4.4, och inledde en andra våg. Under 2014–2021 samexisterade många olika genotyper och spreds genom både vildfåglar och domesticerade flockar, särskilt i Europa, Asien och senare i Amerika. En tredje våg började omkring 2021 med framväxten av klad 2.3.4.4b H5N1, som etablerade sig i flera regioner och orsakade utbrott året runt — ett ”endemiskt” mönster snarare än tillfälliga vintersprång. 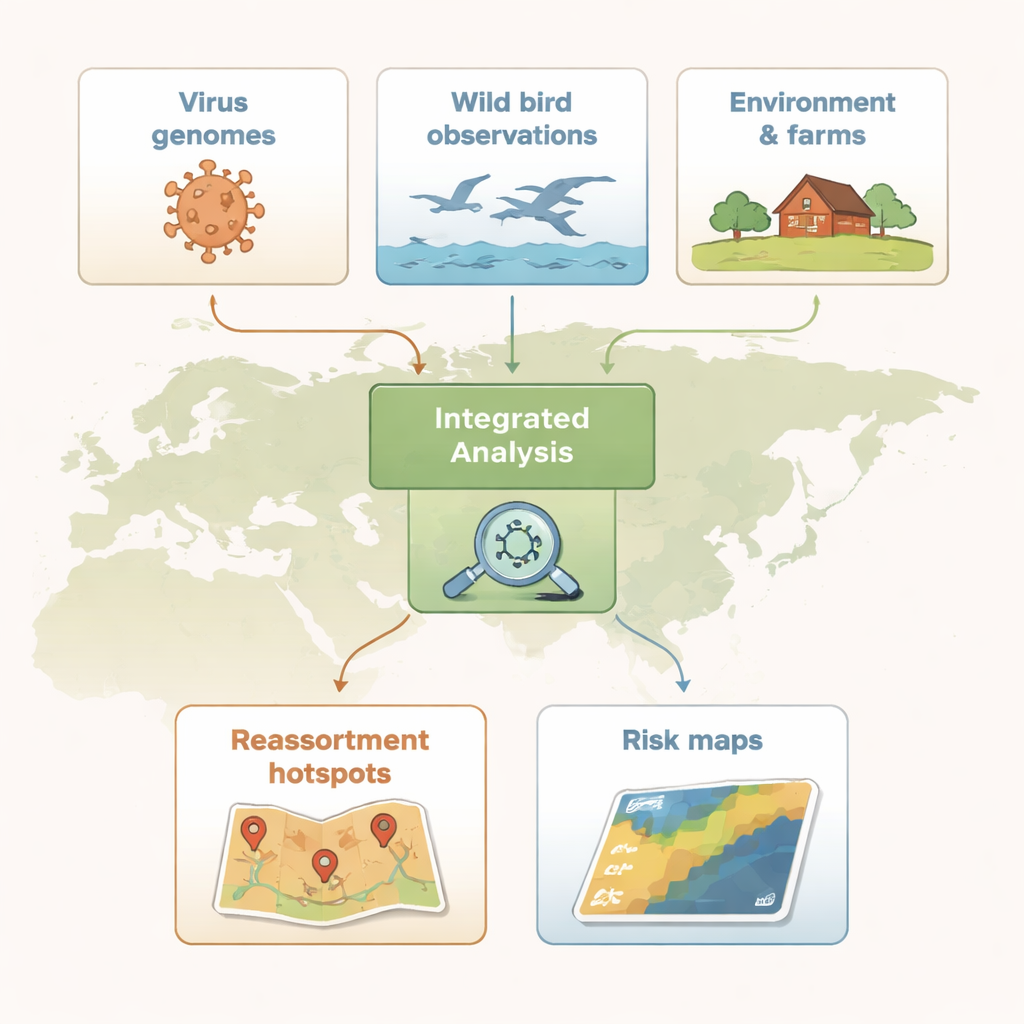
Kartlägga dolda hotspot
För att lokalisera var genutbytet var mest intensivt delade forskarna upp Eurasien i 100-kilometersrutor och räknade hur många olika H5-genotyper som upptäcktes i varje ruta. Med en rumslig statistik som framhäver kluster identifierade de reassorteringshotspots — områden där många genotyper förekom tillsammans oftare än väntat. I början var dessa hotspots koncentrerade i Sydostasien. I den andra vågen förflyttade de sig norrut och västerut, och dök upp längs Stilla havets kuster i Östasien och över Centraleuropa och Västeuropa, inklusive delar av Danmark, södra Sverige och norra Italien. Dessa mönster antydde att både geografi och jordbruksmetoder styrde virusets evolution.
Fågelsamhällen, gårdar och miljön
Hotspots uppstår inte från en enda ”dålig” fågelart eller en typ av gård; de utvecklas där många faktorer överlappar. Teamet kombinerade citizen science-observationer från eBird-projektet med markanvändningskartor, data om fjäderfädtäthet och register över H5-utbrott i gårdar. De identifierade först vildfågelsarter som tenderade att finnas i hotspot-rutor, med fokus på tre stora fågelordningar: vattenfåglar såsom änder och gäss (Anseriformes), strandfåglar (Charadriiformes) och tättingar (Passeriformes). Överraskande nog hade många högriskarter aldrig formellt testats för fågelinfluensa. För att fånga den kombinerade effekten av flera arter byggde författarna en ”polyspecies risk score” som sammanfattade hur sannolikt ett områdes fågelsamhälle var att stödja reassortering. De lade sedan till information om kyckling- och andtäthet, gårdsutbrott och marktyper som åkermark eller bebyggda områden för att uppskatta vilka kombinationer av förhållanden som starkast förutsade hotspots.
Från våtmarker till hönshus
Analysen visade på ett skifte i virusets ekologiska nisch. Under de tidiga åren var reassortering främst kopplat till anduppfödning, vilket stämmer med bilden av ankor som en tyst reservoar som bär viruset utan uppenbar sjukdom. Med tiden, när högpatogena H5-virus blev etablerade på hönsgårdar — understödda i vissa regioner av långvarig cirkulation och vaccinationspraxis — flyttade de starkaste signalerna mot områden med hög kycklingtäthet och blandade jordbruksmiljöer. Bebyggda områden i delar av Asien och åkermark i Europa korrelerade också med hotspots, vilket sannolikt speglar där människor, gårdar och vildfåglar möts. Samtidigt framträdde icke-vattenfåglar som tättingar, som finns i stora antal kring fält, förorter och lador, i allt större utsträckning som en brygga mellan vilda habitat och fjäderfästall.
Vad detta betyder för beredskap
För icke-specialister är huvudbudskapet att farliga nya former av H5-fågelinfluensa troligen uppstår där tät fjäderfäuppfödning, artrika vildfågelsamhällen och människopåverkade landskap möts. Genom att slå samman genetiska data, fågelobservationer och miljöinformation i enhetliga riskkartor erbjuder denna studie en vägledning för var övervakning kan vara mest effektiv — vare sig det innebär att testa underutforskade fågelgrupper, skärpa biosäkerheten kring högriskgårdar eller övervaka regioner där viruset blivit endemiskt. Att förstå och bevaka dessa genetiska ”remixzoner” är ett praktiskt steg för att minska risken att ett djurvirus överraskar oss med ytterligare ett språng i utbredning, virulens eller värdarter.
Citering: Chen, BJ., Liang, CC., Li, YT. et al. Integrated approaches to explore temporal-spatial changes in gene reassortment of highly pathogenic avian influenza A(H5) virus in Eurasia, 2000–2023. Sci Rep 16, 7518 (2026). https://doi.org/10.1038/s41598-026-38466-y
Nyckelord: fågelinfluensa, H5N1, vildfåglar, fjäderfäuppfödning, virusets evolution