Clear Sky Science · sv
Maskininlärningsinteratomär potential för de strukturella egenskaperna hos järnoxider
Varför rostiga berg spelar roll
Järnoxider – mineralen som ger rost dess färg – stöder tyst mycket av det moderna livet. De är huvudkällan till järn för stålproduktion, viktiga komponenter i batterier och solceller och hjälper till att rena förorenat vatten. Trots deras betydelse har vi dock svårt att förutsäga hur dessa material beter sig under verkliga förhållanden, särskilt på atomnivå. Den här artikeln beskriver hur forskare använde modern artificiell intelligens för att bygga en snabb och precis digital modell av en viktig järnoxid, hematit, vilket öppnar dörren för mer tillförlitliga virtuella experiment inom allt från malmhantering till rena energitekniker.
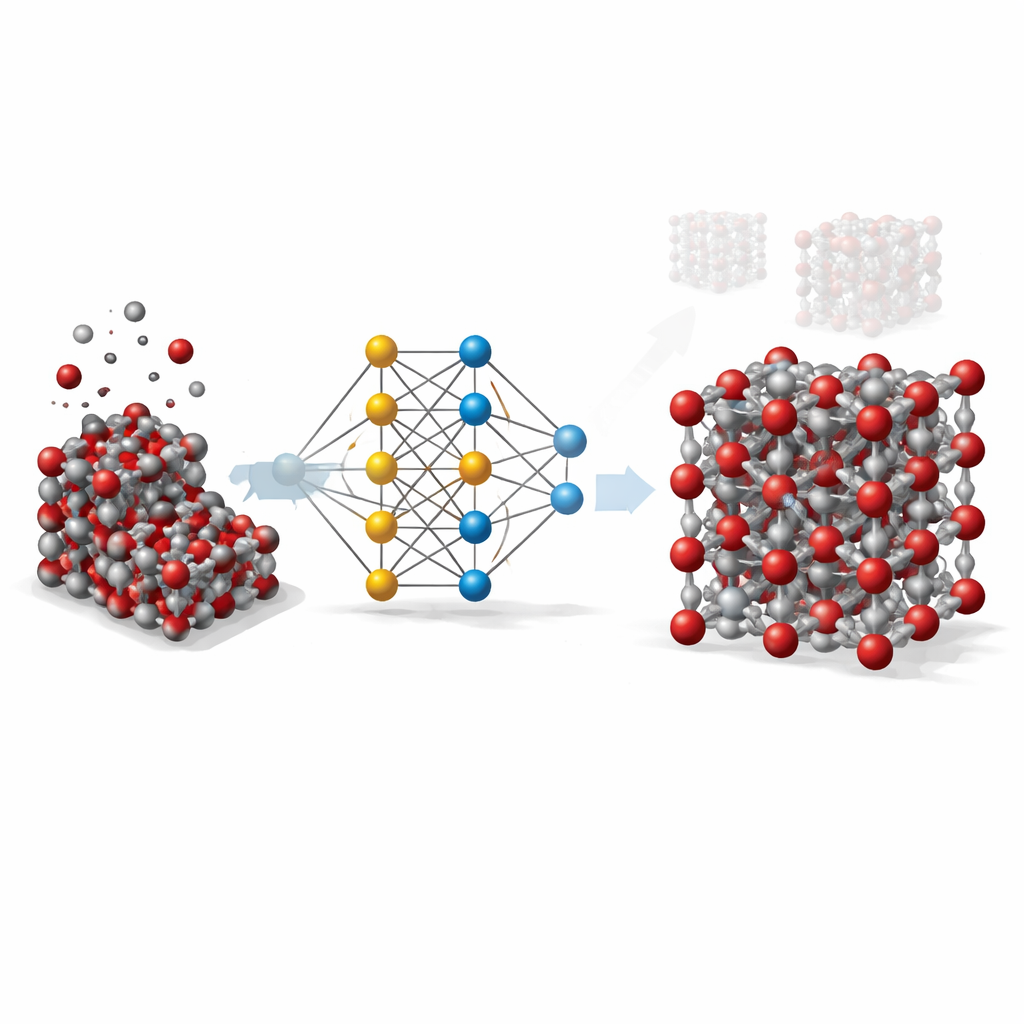
Från kostsamma beräkningar till smarta genvägar
För att förstå ett fast ämne som hematit i detalj förlitar sig forskare i idealfallet på kvantmekaniska metoder som följer hur elektroner och atomer påverkar varandra. Dessa metoder är mycket precisa men så beräkningsintensiva att de är opraktiska för simuleringar av stora prover eller långa tidsskala. Klassiska modeller är däremot snabba men förenklade: de bygger på enkla formler anpassade för specifika situationer och misslyckas ofta när temperatur, tryck eller kristallformer förändras. Det arbete som presenteras här syftar till att överbrygga detta gap genom att använda maskininlärning för att efterlikna kvantberäkningarnas noggrannhet samtidigt som hastigheten hos traditionella modeller behålls.
Lära ett neuralt nätverk om atomer
Teamet byggde vad som kallas en grafneuralt nätverkspotential för hematit. I detta tillvägagångssätt behandlas varje atom som en nod i ett nätverk, och bindningar och närliggande atomer är förbindelserna mellan noderna. För att lära detta nätverk hur atomer i hematit drar i och påverkar varandra genererade forskarna först tusentals atomära ögonblicksbilder med standardiserade simuleringar över ett brett spektrum av temperaturer, tryck och kristalldistorsioner, inklusive både bulkkristaller och exponerade ytor. De använde sedan en avancerad kvantmetod (DFT+U) för att beräkna energi, krafter och interna spänningar för varje ögonblicksbild, och tränade det neurala nätverket att reproducera dessa värden så nära som möjligt.
Kontrollera modellen mot verkligheten
När den var tränad testades den nya potentialen – kallad Fe-MLIP – noggrant. Författarna jämförde dess förutsägelser för grundläggande strukturella storheter såsom gitterdimensioner och hur kristallen töjs under belastning med både experiment och flera allmänt använda klassiska modeller. Fe-MLIP återgav den kända kristallstrukturen hos hematit inom några procent och fångade dess elastiska beteende nästan lika väl som direkta kvantberäkningar, och överträffade tydligt andra kraftfält för många egenskaper. Den presterade också väl i mer subtila tester, såsom hur materialet utvidgas med temperaturen och hur dess atomer vibrerar, vilket är viktigt för värmeledning och spektroskopi. Dessa vibrationsfrekvenser, som aldrig visades explicit under träningen, låg närmare uppmätta värden än de från konkurrerande modeller.
Sträcka sig bortom ett enda mineral
Forskarna utforskade sedan hur långt denna hematitbaserade modell kunde sträckas. De tillämpade den på närbesläktade järnoxider – maghemit och magnetit – som delar liknande atomära byggstenar men skiljer sig i kristallarrangemang och järnens laddningstillstånd. Även om Fe-MLIP inte tränades på dessa faser gav den rimliga värden för deras gitterstorlekar och styvhet, ofta med lika goda eller bättre resultat än specialiserade klassiska modeller. Potentialen fångade även den relativa stabiliteten hos viktiga kristallytor och till och med trender i energikostnaden för att skapa atomvakansier, egenskaper som är avgörande för att förstå korrosion, katalys och batteriprestanda.
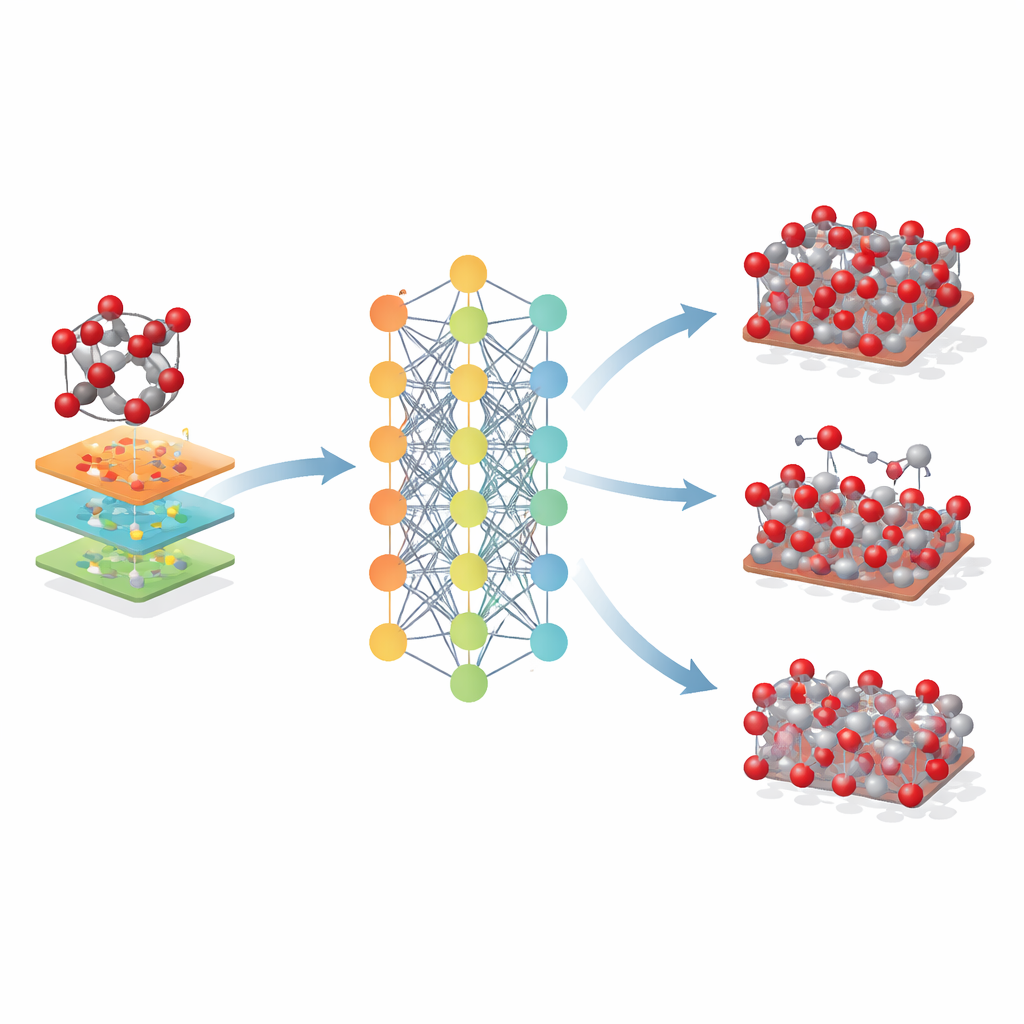
Vad detta betyder för framtida materialdesign
För icke-specialister är slutsatsen att detta arbete ger en kraftfull ny ”digital tvilling” för järnoxider. Fe-MLIP-modellen låter forskare köra stora, långvariga simuleringar av hematit och relaterade material med nästan kvantnivåns tillförlitlighet men till en bråkdel av kostnaden. Medan den ärver vissa begränsningar från den underliggande kvantmetoden och för närvarande är fokuserad på järn och syre, möjliggör den redan mer realistiska studier av hur dessa mineral reagerar på påfrestning, värme, ytor och defekter. I praktiska termer kan ett sådant verktyg påskynda utvecklingen av bättre stålframställningsprocesser, mer effektiva katalysatorer och batterier samt förbättrade miljötekniker som förlitar sig på järnoxider – allt genom att låta forskare testa idéer på datorn innan de går vidare till labbet eller gruvan.
Citering: Torres, A., de Oliveira, A.B., Barbosa, M.d.S. et al. Machine learning interatomic potential for the structural properties of iron oxides. Sci Rep 16, 8576 (2026). https://doi.org/10.1038/s41598-026-38096-4
Nyckelord: hematit, järnoxider, maskininlärningspotential, grafneurala nätverk, molekyldynamik