Clear Sky Science · sv
Molekylär karaktärisering av recessivt ärftliga ataktiska och neuropatiska sjukdomar i konsanguina pakistanska familjer
Varför detta är viktigt för familjer och läkare
Problem med balans, gång och känsel i händer och fötter kan vara djupt funktionsnedsättande, särskilt när de börjar i barndomen och gradvis förvärras under livet. För många familjer, särskilt i regioner där kusiner ofta gifter sig, går dessa symtom i flera generationer utan en tydlig förklaring. Denna studie tar sig an en angelägen fråga för sådana familjer i Pakistan: kan modern DNA‑analys slutligen avslöja de dolda genetiska orsakerna till deras ataxi (dålig balans och koordination) och perifera neuropati (skada på nerverna i extremiteterna), och hjälpa läkare att erbjuda klarare diagnoser och potentiella behandlingar?
Spåra ärftlig sjukdom genom stora familjer
Forskarna arbetade med sju omfattande pakistanska familjer där flera medlemmar hade allvarliga rörelse‑ och nervproblem. Vissa personer hade främst ataxi, vilket försvårade stadigt gångsätt eller kontroll av tal och ögonrörelser. Andra uppvisade klassiska tecken på perifer neuropati, såsom muskelförtvining i händer och fötter, fotdeformiteter och förlust av reflexer. I dessa familjer var föräldrarna släkt med varandra, vilket ökade chansen att barn skulle ärva två kopior av samma ovanliga defekta gen. Med blodprov från drabbade och opåverkade släktingar genomförde teamet exomsekvensering — att läsa nästan alla protein‑kodande delar av genomet — för att söka efter skadliga förändringar som följde sjukdomen genom familjeträden. 
Uppspårning av sällsynta felande gener
Genom att filtrera bort vanliga och ofarliga DNA‑skillnader fann forskarna sannolika sjukdomsframkallande varianter i fem av de sju familjerna. Var och en av dessa familjer bar på sin egen specifika genetiska förändring, och alla följde ett recessivt mönster: personer blev sjuka endast när de ärvde två defekta kopior, en från vardera föräldern. I en familj med balansproblem i vuxen ålder och talrubbningar var boven en sällsynt förändring i genen MFSD8, som hjälper till att transportera material till cellens återvinningskompartiment, lysosomer. I en annan familj kopplades en skadlig förändring i AFG3L2, ett protein som upprätthåller mitokondriernas hälsa — cellens kraftverk — till barndomsdebuterad spastisk ataxi med dystoni (onormala muskelkontraktioner). En tredje familj bar på ett frameshift‑fel i SETX, en gen som skyddar DNA vid reparation och som redan är känd för att orsaka ataxi med okulomotorisk apraxi, en störning som också påverkar ögonrörelser.
En närmare titt på ärftlig nervskada
Ytterligare två familjer hade en form av Charcot‑Marie‑Tooth (CMT), en grupp ärftliga tillstånd som skadar de långa nerverna till fötter och händer. I båda fann forskarna skadliga varianter i genen GDAP1, som är avgörande för normal mitokondriell funktion i nervceller. En GDAP1‑förändring kapade proteinet och var associerad med mycket svår, tidigt debuterande sjukdom; en annan bytte ut ett enda byggsten i proteinet och gav en något mildare förlopp. Slående nog var den mest svårt drabbade patienten i en CMT‑familj också homozygot för en känd sjukdomsvariant i en andra gen, MMACHC, som är involverad i vitamin B12‑metabolismen och ibland kan behandlas med vitaminbaserade terapier. Denna dubbla påverkan kan förklara varför hans symtom var värre än hos släktingar som saknade MMACHC‑varianten.
När DNA‑sökningen inte räcker
Inte varje familj gav ett tydligt genetiskt svar. I två av de sju familjerna kunde teamet inte hitta någon enskild förändring i exomet som övertygande matchade sjukdomsmönstret. I ett fall identifierade de en variant i genen EHHADH som följde arvsmönstret men som förutses vara ofarlig och som är känd för att orsaka en annan njurrelaterad åkomma när den är förändrad. I ett annat fall visade sig två kusiner med liknande rörelseproblem ha olika bakomliggande orsaker: en pojke bar på en känd skadlig variant i ALS2, som kan leda till juvenil form av motorneuronsjukdom, medan hans drabbade kusiner inte gjorde det. Dessa olösta fall tyder på att viktiga mutationer kan ligga i delar av genomet som standard exomsekvensering inte fångar upp, eller att mer än en subtil genetisk faktor kan samverka.
Vad detta betyder för patienter och framtida vård
Tillsammans visar resultaten att kraftfulla DNA‑verktyg kan avslöja de specifika generna bakom komplexa nerv‑ och balansstörningar, även i miljöer med begränsade resurser. För de fem familjerna med tydliga fynd omvandlar arbetet vaga etiketter som ”ataxi” eller ”neuropati” till precisa diagnoser kopplade till särskilda gener, vilket kan vägleda genetisk rådgivning, informera prognos och i vissa fall peka på behandlingsalternativ, såsom vitamin B12‑relaterade terapier för MMACHC‑associerade sjukdomar. Studien breddar också forskarnas förståelse för hur gener som MFSD8, AFG3L2, SETX, GDAP1, MMACHC och ALS2 formar nervcellernas hälsa i hjärna, ryggmärg och perifera nerver. Framöver kommer mer omfattande genomsekvensering och funktionella studier att behövas för att lösa de återstående mysterierna och för att omvandla dessa genetiska insikter till tidigare diagnos och bättre vård för drabbade barn och vuxna. 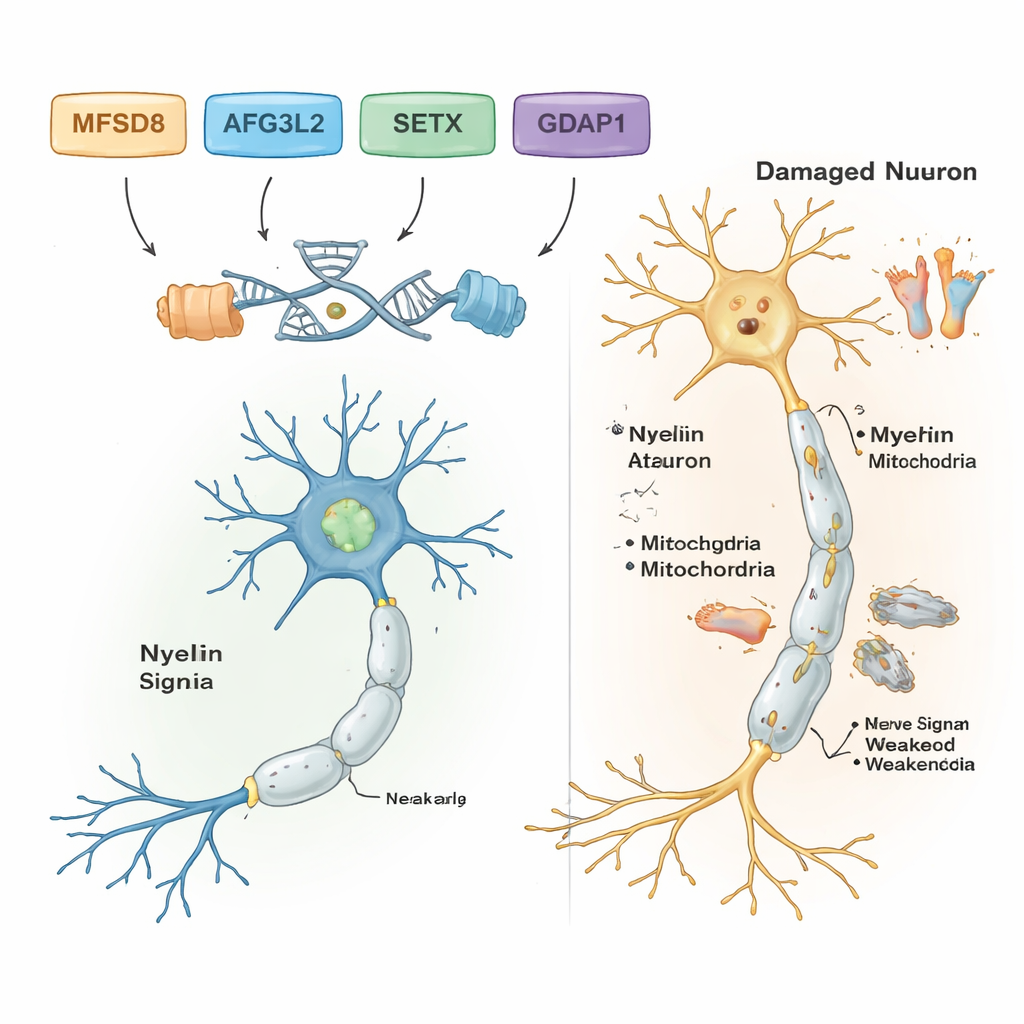
Citering: Aslam, F., Wajid, M., Butt, A.I. et al. Molecular characterization of recessively inherited ataxic and neuropathic disorders in consanguineous Pakistani families. Sci Rep 16, 6529 (2026). https://doi.org/10.1038/s41598-026-37808-0
Nyckelord: ataxi, perifer neuropati, exomsekvensering, Charcot-Marie-Tooth-sjukdom, genetisk diagnos