Clear Sky Science · sv
Optimera urval av funktioner i cancer‑mikroarraydata med ett högdrivet evolutionärt ramverk för högdimensionella utrymmen
Varför valet av rätt gener är viktigt
Cancertester baserade på moderna gentekniker kan mäta tiotusentals gener samtidigt, men läkare har ofta data från bara några dussin patienter. Dold i denna enorma ”gen‑djungel” finns ett mycket mindre antal signaler som verkligen skiljer en cancertyp från en annan, eller en tumör från frisk vävnad. Den här artikeln presenterar en ny smart sökmetod för att automatiskt plocka ut dessa nyckelgener, med målet att göra datorstödd cancerdiagnostik mer exakt, snabbare och enklare att tolka.
För många signaler, för lite data
Mikroarrayexperiment och liknande tekniker gör det möjligt för forskare att mäta aktivitetsnivåer för tusentals gener i varje patientprov. Men antalet prover är vanligtvis mycket litet, ibland färre än hundra. Många av dessa genavläsningar är brusiga, redundanta eller irrelevanta för den aktuella sjukdomen. Att behålla alla kan överväldiga inlärningsalgoritmer, göra beräkningar långsamma och ge vilseledande modeller som hakar upp sig på slumpmässiga egenheter istället för verklig biologi. Processen att skära ner detta till en användbar delmängd kallas ”funktionsurval”, och den är avgörande om vi vill ha tillförlitliga prediktioner från högdimensionella medicinska data.
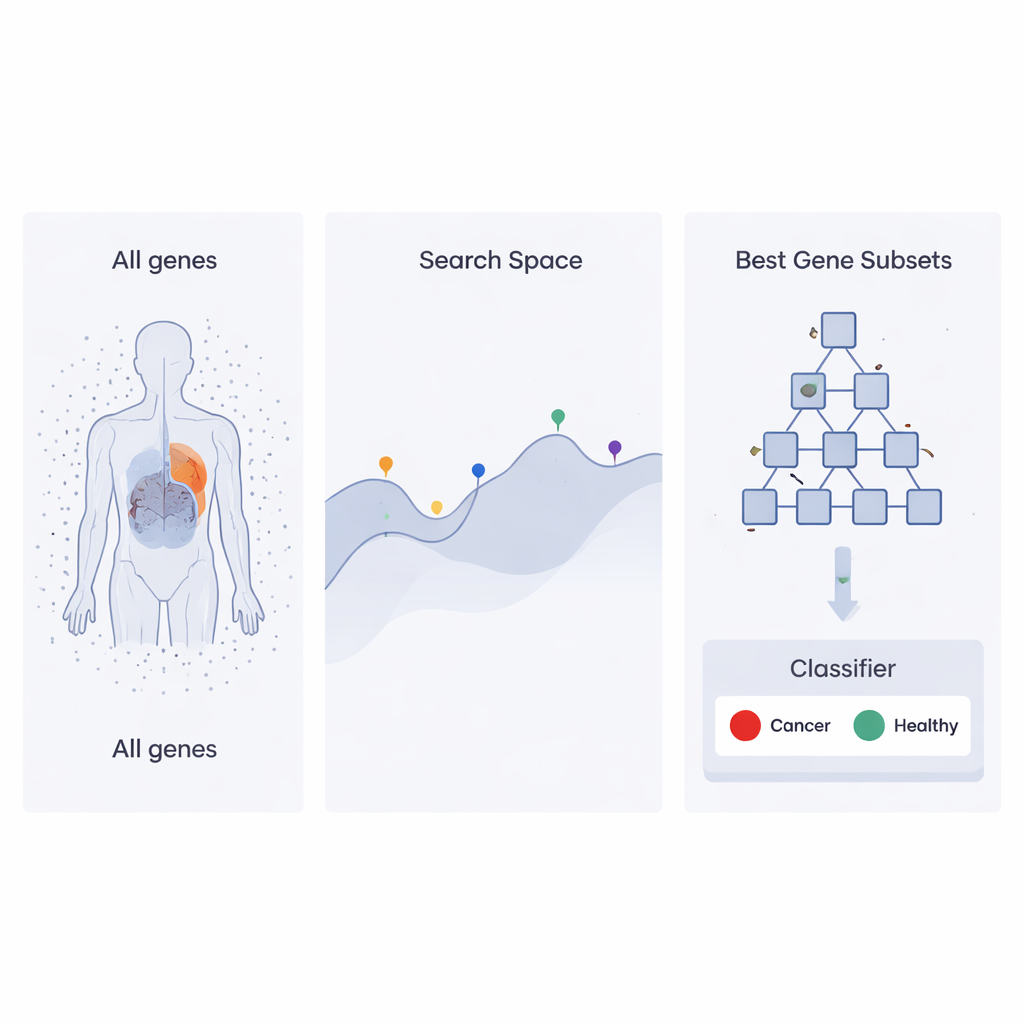
En sökstrategi inspirerad av företagsstegar
Författarna bygger vidare på en nyligen utvecklad optimeringsmetod kallad Heap‑Based Optimizer (HBO), som lånar idéer från hur anställda är organiserade i ett företag. Föreställ dig varje möjlig uppsättning gener som en ”anställd” vars arbetsinsats bedöms efter hur väl den hjälper en klassificerare att skilja cancertyper från friska prover. Dessa anställda ordnas i en hierarki, som en företagsstege, med hjälp av en datastruktur känd som en heap. Högpresterande gensatser sitter nära toppen, medan svagare sitter längre ner. Över många omgångar justerar de lägre rankade sina val genom att kopiera och lätt modifiera vad deras chefer och kollegor gör, och driver gradvis hela organisationen mot bättre lösningar.
Från råa genavläsningar till skarpare mönster
För att göra sökningen mer effektiv förlitar sig inte författarna enbart på råa genavläsningar. De omformar först mikroarraydata till ett bildliknande format och tillämpar en teknik kallad Histogram of Oriented Gradients (HOG), som är mycket använd i datorseende. HOG fångar hur uttrycksnivåer förändras över gener och framhäver lokala mönster snarare än isolerade mätpunkter. Dessa mönsterbaserade funktioner kombineras sedan med den ursprungliga geninformationen. En enkel klassificerare kallad k‑Nearest Neighbors (KNN) fungerar som ”domare” och poängsätter varje kandidatdelmängd av gener efter hur exakt den klassificerar nya prover samtidigt som den belönar mindre, mer kompakta uppsättningar.
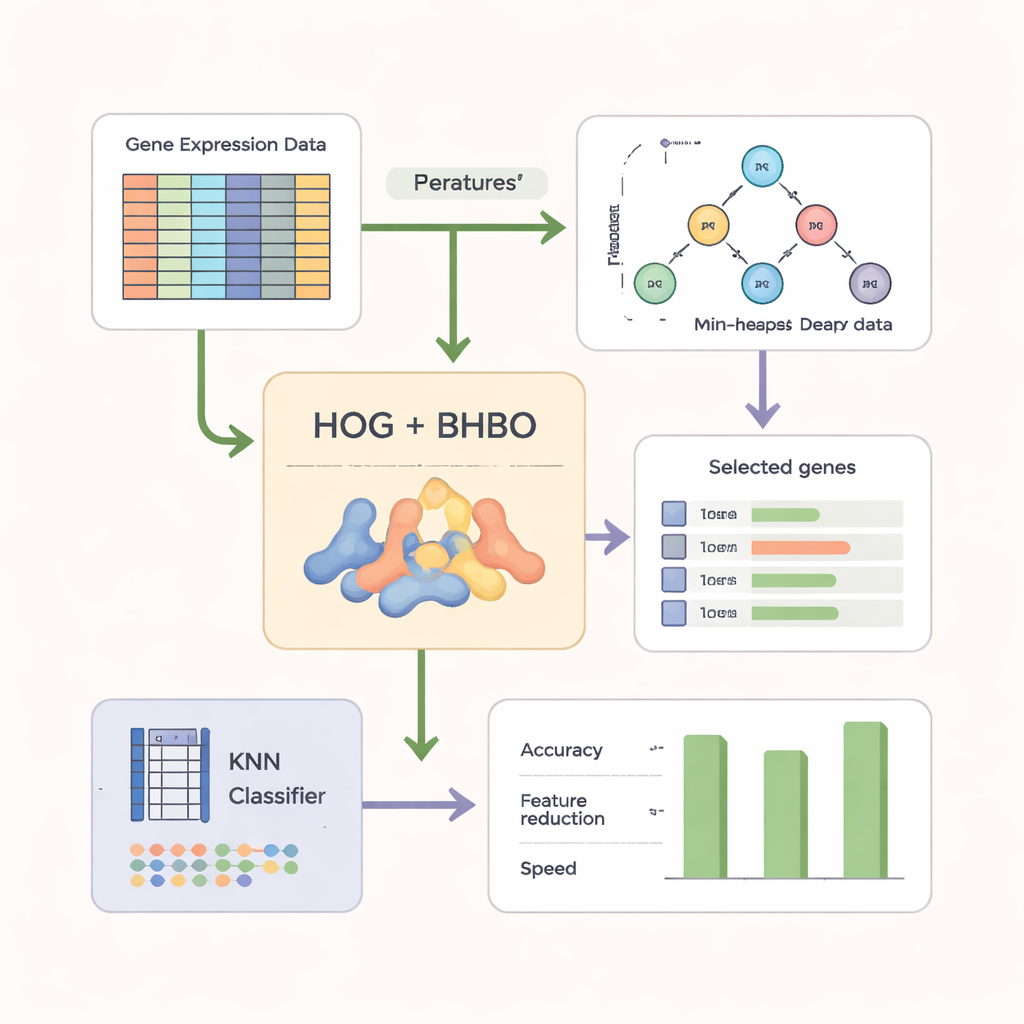
Testning på flera cancer‑datamängder
Forskarna utvärderade sin binära version av Heap‑Based Optimizer (BHBO) på nio publika cancer‑mikroarraydatamängder, inklusive hjärntumörer, leukemier, prostatacancer och blandade tumörsamlingar med många undertyper. Varje datamängd hade från tusentals upp till över femton tusen mätta gener men relativt få patientprover. För varje datamängd kördes BHBO upprepade gånger och jämfördes med sju välkända sökmetoder, såsom genetiska algoritmer och partikel‑svärmsoptimering. Teamet mätte inte bara noggrannhet, utan också hur många gener som behölls, hur snabbt sökningen konvergerade och hur stabila resultaten var när data stördes av simulerat brus, batch‑effekter och felaktiga etiketter.
Vad den nya metoden uppnådde
Över de nio datamängderna nådde den högdrivna metoden en genomsnittlig klassificeringsnoggrannhet på cirka 95 procent samtidigt som antalet gener reducerades med mer än 85 procent. Den slog tydligt konkurrerande metoder på flera datamängder och visade snabbare konvergens, vilket innebär att den hittade bra gensatser på färre söksteg. Även när författarna avsiktligt korruptade data — genom att lägga till brus eller byta ut några provetiketter — sjönk metodens prestanda endast marginellt och förblev bättre än alternativen. Statistiska tester bekräftade att dessa vinster sannolikt inte berodde på slumpen.
Vad detta betyder för framtida cancerdiagnostik
I praktiska termer visar detta arbete att en noggrant utformad sökstrategi kan sålla igenom enorma genetiska datamängder och upptäcka små, informationsrika paneler av gener som ändå klassificerar cancer mycket väl. För kliniker och forskare är sådana kompakta genset lättare att biologiskt validera, billigare att mäta i uppföljande tester och mer lämpade för integrering i beslutsstödsverktyg. Medan metoden inte direkt upptäcker nya läkemedel eller signalvägar, riktar den strålkastarljuset mot lovande genetiska markörer och hjälper andra studier att fokusera på de mest informativa signalerna som döljer sig i högdimensionell cancerdata.
Citering: Alweshah, M., Jebril, H., Kassaymeh, S. et al. Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces. Sci Rep 16, 6726 (2026). https://doi.org/10.1038/s41598-026-37803-5
Nyckelord: cancer mikroarray, funktionsurval, metaheuristisk optimering, gen‑biomarkörer, medicinsk datautvinning