Clear Sky Science · sv
En genoms‑strukturanpassad ram för ROH‑baserad inaveluppskattning i Penaeus vannamei
Varför räkens släktträd spelar roll för din middag
Moderna räkodlingar levererar stora delar av världens skaldjur, men att ständigt avla på samma familjelinjer kan tyst urholka deras hälsa. När nära släktingar får avkomma kan skadliga dolda varianter parar sig, vilket minskar tillväxt, överlevnad och motståndskraft mot sjukdomar. Denna studie ställer en till synes enkel men långtgående fråga för global akvakultur: hur kan vi mäta inavel i odlade räkor tillräckligt noggrant för att hålla bestånden friska och produktiva?
Dolda genetiska spår av inavel
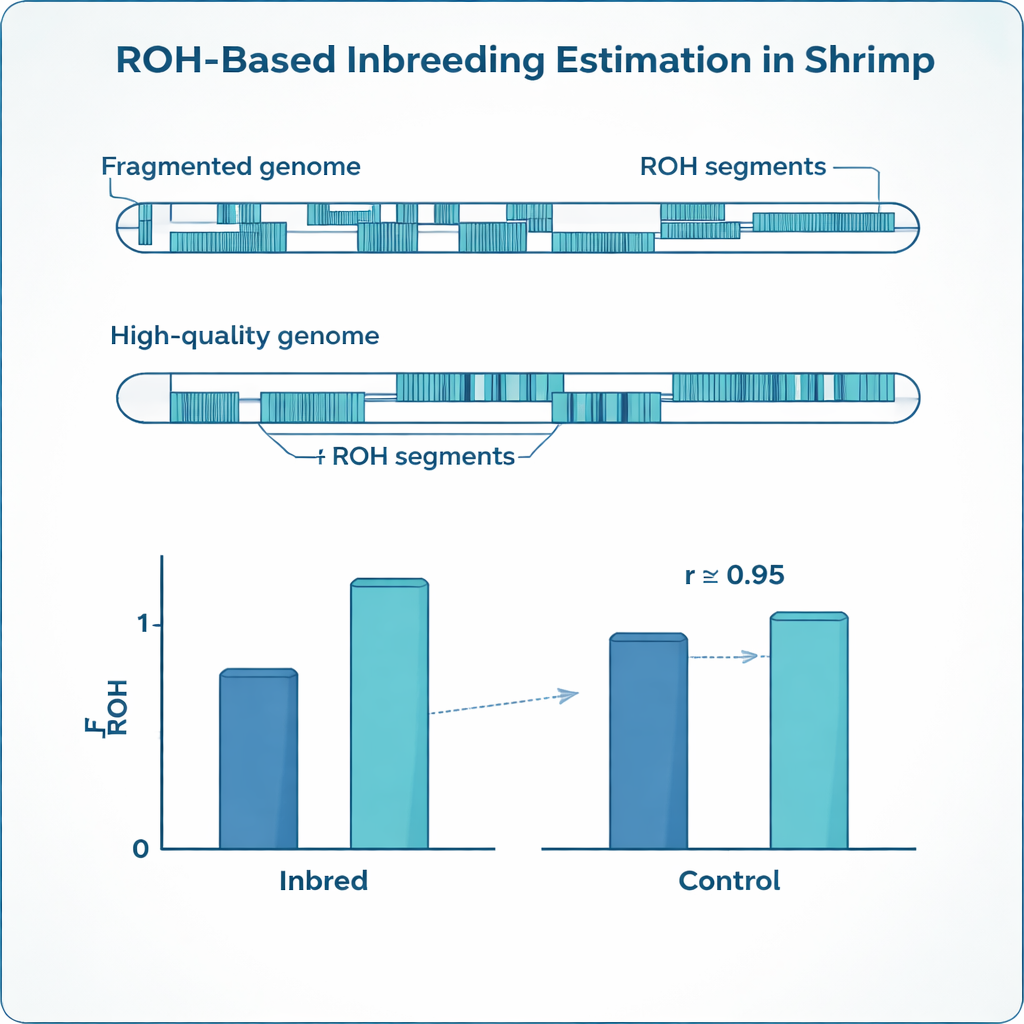
Inavel lämnar igenkännbara avtryck i DNA:t. Istället för att bära två något olika varianter av många gener tenderar inavlade djur att ha långa segment där båda kopiorna är identiska. Genetiker kallar dessa segment för ”runs of homozygosity”, eller ROH. Genom att summera hur stor del av ett djurs genom som faller inom dessa segment kan forskare uppskatta dess inavelsnivå mer precist än genom pappersstamtavlor, som ofta är ofullständiga eller innehåller fel. Denna ROH‑baserade måttstock, känd som FROH, har blivit standard inom nötkreatur, grisar och andra husdjursarter, men räkors genom utgör särskilda utmaningar som gör färdiga metoder opålitliga.
Varför räkgenom är särskilt knepiga
Vitbenad räka (Penaeus vannamei), världens mest odlade räka, har mycket fragmenterade och komplexa genom. Istället för långa, kontinuerliga kromosomsekvenser är många tillgängliga genomkartor brutna i tusentals mindre bitar, separerade av luckor och repetitiva regioner. Genetiska markörer är ojämnt spridda över detta lapptäcke, och arten har mycket hög genetisk mångfald. Metoder och programinställningar som ursprungligen ställts in för ordnade, väl kartlagda däggdjursgenom kan därför missta tekniska luckor för verkliga avbrott i ROH, eller förväxla korta, bakgrundslika sekvenser med verkliga tecken på inavel. Resultatet blir en hög risk för felaktiga uppskattningar av hur inavlad en räka faktiskt är.
Att bygga en genomanpassad mätsticka
För att lösa detta utvecklade författarna en ”genom‑struktur‑anpassad” ram som skräddarsyr ROH‑analys till räkors DNA‑särdrag. De skapade tretton starkt inavlade räkfamiljer genom kontrollerade parningar och sekvenserade djupt genomet hos fem av dessa familjer samt deras föräldrar. Avgörande var att de anpassade samma sekvensdata mot två mycket olika referensgenom: en äldre, fragmenterad sammansättning och en nyare, mycket mer kontinuerlig version. Med det vanliga analysverktyget PLINK testade de systematiskt hur åtta centrala inställningar påverkade ROH‑kallelser, med fokus på tre som var särskilt känsliga för genomstruktur: hur tätt markörer måste ligga, hur stort gap mellan markörer som får vara inom ett run och hur långt ett run måste vara för att räknas. De konstruerade empiriska, icke‑överlappande genomfönster för att följa lokal markörtäthet och saknade data, och använde ”genomisk täckning” av dessa fönster, tillsammans med stabiliteten i FROH och ROH‑längder, som objektiva riktlinjer för att välja rimliga trösklar.
Olika rattar, samma inavelsbild
De optimerade inställningarna visade sig vara mycket olika för de två referensgenomen. Det fragmenterade genomet krävde mycket tätare markörer, kortare tillåtna gap och kortare minimilängd för ett run än det högkvalitativa genomet för att undvika att bryta upp verkliga ROH i många små bitar. Ändå konvergerade inavelsuppskattningarna från båda referenserna efter separat inställning: genomsnittlig FROH i de inavlade räkorna var omkring 0,24 i båda fallen, vilket stämde väl överens med det förväntade värdet från de planerade parningarna och visade stark överensstämmelse sinsemellan. Samtidigt avslöjade det mer kontinuerliga genomet färre men mycket längre ROH‑segment, medan den fragmenterade kartan styckade många av dessa i korta sträckor. Studien visade också slående skillnader mellan helsyskon: även inom samma familj varierade inavelsnivåerna kraftigt, något enkla stamtavlor inte kan fånga.
Skarpare verktyg för friskare räktbestånd
För icke‑specialister är budskapet enkelt: att mäta inavel från DNA kan vara mycket precist i odlade räkor, men endast om metoden tar hänsyn till det underliggande genomets struktur. Genom att tillhandahålla ett praktiskt recept för att ställa in ROH‑analys för fragmenterade kräftdjursgenom gör detta arbete det möjligt för uppfödare att övervaka inavel individ för individ, istället för att förlita sig på ofullkomliga familjeförteckningar. Det kan i sin tur hjälpa till att utforma parningsplaner som bevarar genetisk mångfald samtidigt som tillväxt och motståndskraft förbättras, vilket stödjer mer hållbar räkproduktion och erbjuder en mall för andra akvakultursarter med liknande genomiska hinder.
Citering: Zou, X., Zhou, H., Liu, M. et al. A genome-structure adaptive framework for ROH-based inbreeding estimation in Penaeus vannamei. Sci Rep 16, 6769 (2026). https://doi.org/10.1038/s41598-026-37622-8
Nyckelord: räkavel, inavel, genomisk selektion, runs of homozygosity, genetik för akvakultur